Choroba Creutzfeldta–Jakoba (CJD) — przegląd i znaczenie
CJD to rzadka, szybko postępująca i śmiertelna choroba prionowa mózgu. Artykuł przedstawia mechanizm prionów, typy choroby, objawy, diagnostykę oraz zasady zapobiegania i kontroli zakażeń.
Przegląd
Choroba Creutzfeldta–Jakoba (CJD) jest rzadką chorobą neurologiczną, należącą do grupy schorzeń prionowych. Ma charakter zwyrodnieniowy, przebiega szybko i ostatecznie prowadzi do śmierci. W literaturze medycznej rozróżnia się kilka postaci CJD, w tym postać sporadyczną, rodzinną, jatrogeniczną oraz związaną z przeniesieniem czynnika zakaźnego z bydła (BSE), zwaną wariantową CJD.
Galeria obrazów
6 Obrazy




Priony i mechanizm choroby
CJD jest wywoływana przez czynnik zakaźny zwany prionem. Priony to patologiczne formy białka, które przyjmują nieprawidłowe konformacje i potrafią przemieniać normalne białka w formy chorobotwórcze. W odróżnieniu od wirusów czy bakterii, priony nie zawierają kwasów nukleinowych i są niezwykle odporne na standardowe metody sterylizacji, co utrudnia kontrolę zakażeń.
Objawy, zmiany morfologiczne i rozpoznanie
Choroba atakuje przede wszystkim mózg i powoduje postępujące uszkodzenie tkanki nerwowej. Typowe objawy obejmują szybko postępującą demencję, mimowolne krótkie skurcze mięśni (mioklonie), zaburzenia chodu i koordynacji, problemy wzrokowe oraz zmiany zachowania. W zaawansowanym stadium może wystąpić stan akinezy z mutyzmem.
- Objawy początkowe: zaburzenia pamięci, trudności w koncentracji, labilność emocjonalna.
- Objawy późne: ciężka demencja, utrata samodzielności, zaburzenia oddychania.
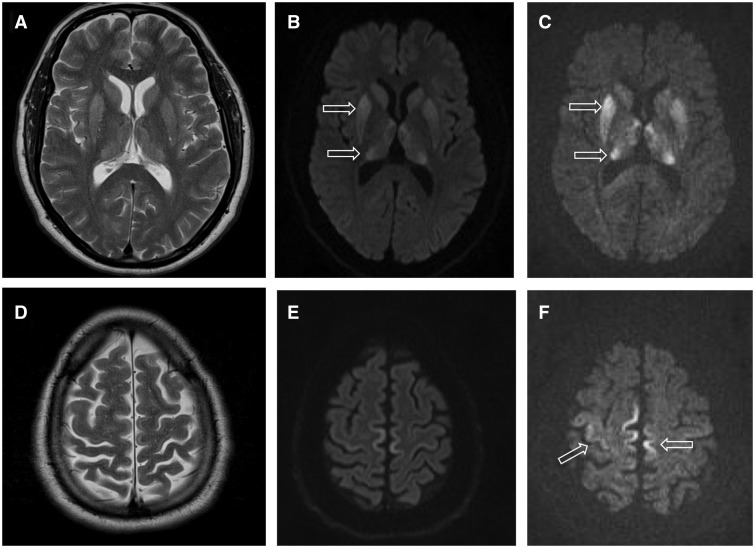
W badaniach anatomicznych mózg chorych przyjmuje strukturę przypominającą gąbkę — widoczne są drobne ubytki i zmiany spongiformne. Zmiana ta jest określana jako tekstura przypominająca gąbkę. Rozpoznanie opiera się na obrazie klinicznym, badaniach neuroobrazowych (MRI), zapisie EEG oraz testach na markery w płynie mózgowo-rdzeniowym; nowoczesne metody laboratoryjne, jak sekwencjonowanie i testy amplifikacji prionów (RT-QuIC), zwiększają precyzję diagnozy.
Historia, typy i znaczenie epidemiologiczne
Nazwę choroba otrzymała od niemieckich neurologów Crautzfeldta i Jakoba, którzy opisali przypadki na początku XX wieku. Istotnym momentem w badaniach nad prionami było sformułowanie hipotezy białkowej oraz przyznanie Nagrody Nobla za odkrycie prionowego mechanizmu chorobowego. W latach 80. i 90. XX wieku powiązano epidemię BSE u bydła z pojawieniem się wariantowej CJD u ludzi, co spowodowało zmiany w przemyśle spożywczym i procedurach bezpieczeństwa.
Zapobieganie, kontrola i leczenie
Nie istnieje skuteczne leczenie przyczynowe CJD; opieka ma charakter paliatywny i wspomagający. Zapobieganie polega na ograniczeniu ryzyka transmisji: ścisłe procedury sterylizacji i dekontaminacji narzędzi chirurgicznych, kontrola przeszczepów tkanek i preparatów biologicznych oraz nadzór nad łańcuchem żywnościowym. Ze względu na odporność prionów na konwencjonalne metody oczyszczania, wymagane są specjalne protokoły postępowania.
Wyróżniki kliniczne: szybkie pogarszanie funkcji poznawczych i ruchowych, brak odpowiedzi na standardowe terapie zakaźne oraz charakterystyczne zmiany neuropatologiczne. Zrozumienie mechanizmów prionowych ma duże znaczenie dla badań nad chorobami neurodegeneracyjnymi i bezpieczeństwem zdrowia publicznego.
Więcej informacji można znaleźć w źródłach specjalistycznych: neurologia, choroby zwyrodnieniowe, BSE, gąbczasta encefalopatia, czynnik zakaźny, prion, białka, tkanka, mózg, tekstura, gąbka.
Rodzaje i przyczyny CJD
Rodzaje CJD obejmują:
- wariant (vCJD):
Ten typ CJD może być spowodowany przez spożycie żywności zawierającej priony, np. mięsa krów chorych na BSE ("choroba szalonych krów"). Jest to jednak bardzo rzadka przyczyna CJD.
- sporadyczne (sCJD):
Jest to najczęstszy typ CJD. 85% przypadków CJD to CJD sporadyczna. Nikt nie wie, co powoduje sCJD; wydaje się, że dzieje się to losowo.
- rodzinną (fCJD):
Większość z pozostałych 15% przypadków CJD to CJD rodzinna. Jest to postać CJD występująca w rodzinach.
- jatrogenny:
Ta forma CJD jest zwykle spowodowana procedurą medyczną, w której dana osoba otrzymuje krew lub tkankę od osoby chorej na CJD. Na przykład, osoba może zachorować na jatrogenną postać CJD, jeśli otrzyma transfuzję krwi lub przeszczep rogówki od osoby chorej na CJD.
Oznaki i objawy
Pierwszym objawem CJD jest demencja, która bardzo szybko się pogłębia. demencja powoduje utratę pamięci, zmiany osobowości i halucynacje.
Inne częste objawy psychiczne obejmują:
- Lęk
- Depresja
- Paranoja
- Objawy obsesyjno-kompulsywne
- Psychoza
Fizyczne objawy CJD często obejmują:
- Kłopoty z mówieniem
- Ruchy szarpane (mioklonie)
- Problemy z utrzymaniem równowagi (ataksja)
- Problemy z chodzeniem
- Trzęsienie się lub sztywność
- Problemy z widzeniem
- Problemy z połykaniem, które mogą utrudniać lub uniemożliwiać jedzenie
- Kłopoty z kaszlem, który może powodować zapalenie płuc
- Ruchy, których pacjent nie jest w stanie kontrolować (dyskineza)
Większość osób z CJD umiera w ciągu sześciu miesięcy od wystąpienia pierwszych objawów. Często umierają z powodu zapalenia płuc spowodowanego kłopotami z kaszlem. Około 15% pacjentów przeżywa dwa lub więcej lat. Niektórzy pacjenci żyją 4-5 lat z objawami głównie psychicznymi, dopóki choroba się nie pogorszy i nie spowoduje więcej objawów fizycznych. Kiedy to nastąpi, chorzy zazwyczaj umierają w ciągu roku.
Objawy CJD są spowodowane obumieraniem coraz większej liczby komórek nerwowych w mózgu. Kiedy naukowcy oglądają tkankę mózgową pacjenta z CJD pod mikroskopem, mogą zobaczyć wiele maleńkich otworów, w których obumarły całe obszary komórek nerwowych.
Diagnoza
Lekarze mogą podejrzewać CJD, gdy u danej osoby występują pewne objawy. Na przykład, otępienie zwykle pogłębia się powoli. Demencja, która pogarsza się bardzo szybko jest nietypowa. Wraz z objawami takimi jak szarpane ruchy, objawy te mogą wskazywać na możliwość wystąpienia CJD.
Następnie można wykonać badania, które wykażą, czy dana osoba ma CJD. Testy te obejmują:
- Elektroencefalografia (EEG): Badanie to pokazuje aktywność elektryczną w mózgu. Lekarz często będzie w stanie zaobserwować zmiany w EEG, które są powszechne u osób z CJD. Rodzaj zmian widocznych w badaniu EEG zależy od rodzaju CJD występującego u pacjenta oraz od zaawansowania choroby.
- Nakłucie lędźwiowe (nakłucie rdzenia kręgowego): Ten test umożliwia badanie płynu mózgowo-rdzeniowego (płynu, który otacza mózg i rdzeń kręgowy) w poszukiwaniu specyficznego białka ("białko 14-3-3").
- MRI mózgu: Badanie, które wykorzystuje bardzo silny magnes do robienia zdjęć mózgu.
- Biopsja: Aby wykonać biopsję, chirurg używa igły do pobrania małego fragmentu tkanki z ciała, aby lekarze mogli obejrzeć go pod mikroskopem. vCJD można zdiagnozować za pomocą biopsji migdałków. W przypadku wszystkich innych typów CJD, biopsja mózgu jest jedynym sposobem, aby stwierdzić na pewno, czy dana osoba ma CJD. Ponieważ jednak biopsja mózgu może spowodować jego uszkodzenie, zwykle nie wykonuje się jej, jeśli inne badania wykazały, że dana osoba prawdopodobnie ma CJD.

Leczenie
Na dzień 2016 roku nie ma leczenia, które leczy CJD lub nawet spowalnia jego skutki. Przeprowadza się wiele eksperymentów, aby spróbować znaleźć metody leczenia.
Obecnie jedynymi metodami leczenia CJD są leki, które leczą objawy choroby i pomagają pacjentom czuć się bardziej komfortowo. Na przykład pacjenci, u których występują drgawki, mogą otrzymywać leki przeciwdrgawkowe. Benzodiazepiny mogą sprawić, że szarpnięcia mięśni będą występować rzadziej.
Pacjenci mogą również zdecydować się na zabiegi medyczne mające na celu złagodzenie objawów choroby. Na przykład, CJD może powodować tak duże problemy z połykaniem, że dana osoba nie może jeść. Niektóre osoby z CJD decydują się na założenie rurki do karmienia, kiedy nie mogą już jeść. Jest to rurka, która wchodzi do żołądka, dzięki czemu specjalny płyn może być podawany bezpośrednio do żołądka, aby zapewnić danej osobie odżywianie.
Powiązane strony
- Prion
- Choroba prionowa
- Choroba śmiertelna
Pytania i odpowiedzi
P: Co to jest choroba Creutzfeldta-Jakoba?
A: Choroba Creutzfeldta-Jakoba (CJD) jest chorobą neurologiczną, która jest zwyrodnieniowa, nieuleczalna i zawsze śmiertelna.
P: Czy istnieje lekarstwo na CJD?
O: Nie, na CJD nie ma lekarstwa.
P: Dlaczego CJD jest czasami określana jako ludzka forma "choroby szalonych krów"?
A: CJD jest czasami nazywana ludzką formą "choroby szalonych krów", ponieważ gąbczasta encefalopatia bydła (BSE), która jest przyczyną jednej z rzadkich odmian CJD, jest powszechnie znana jako "choroba szalonych krów".
P: Co jest przyczyną CJD?
O: CJD jest wywoływane przez czynnik zakaźny zwany prionem, który jest białkiem nieprawidłowo złożonym i może tworzyć swoje kopie, zmieniając prawidłowo złożone białka w białka źle złożone.
P: Co się dzieje z tkanką mózgową w CJD?
O: CJD powoduje, że tkanka mózgowa bardzo szybko staje się niezdrowa, w wyniku czego w mózgu powstają dziury i zmienia się jego struktura, która przypomina kuchenną gąbkę.
P: Czy BSE to ta sama choroba co CJD?
O: Nie, BSE nie jest tą samą chorobą, co CJD; w rzeczywistości jest przyczyną jednego z rzadkich typów CJD.
P: W jaki sposób priony wywołują CJD?
O: Priony wywołują CJD poprzez nieprawidłowe składanie i tworzenie swoich kopii kosztem prawidłowo złożonych białek w mózgu. W wyniku tego dochodzi do zniszczenia zdrowej tkanki mózgowej i powstania charakterystycznych dla tej choroby otworów.
Powiązane artykuły
Autor
AlegsaOnline.com Choroba Creutzfeldta–Jakoba (CJD) — przegląd i znaczenie Leandro Alegsa
URL: https://pl.alegsaonline.com/art/24152
Źródła
- cdc.gov : "CJD (Creutzfeldt–Jakob Disease, Classic)"
- ncbi.nlm.nih.gov : "Creutzfeldt–Jakob disease: Transmissible spongiform encephalopathy; vCJD; CJD; Jacob-Creutzfeldt disease"
- bmj.com : "Bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease"
- doi.org : 10.1101/SQB.1996.061.01.052
- pubmed.ncbi.nlm.nih.gov : 9246478
- www3.interscience.wiley.com : "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course"
- doi.org : 10.1111/j.1440-1789.2008.00904.x
- pubmed.ncbi.nlm.nih.gov : 18410280
- who.int : who.int: "Fact sheets no 180: Variant Creutzfeldt-Jakob disease" Feb 2012 ed.
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- doi.org : 10.1056/NEJM198610163151605
- pubmed.ncbi.nlm.nih.gov : 3762620
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- merckmanuals.com : "Creutzfeldt–Jakob Disease (CJD)"
- ncbi.nlm.nih.gov : "Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy"