Mukowiscydoza — definicja, przyczyny, objawy i leczenie
Mukowiscydoza — przejrzyste wyjaśnienie przyczyn, objawów i leczenia: genetyka, diagnostyka, wpływ na płuca i układ pokarmowy oraz aktualne terapie i porady dla pacjentów.
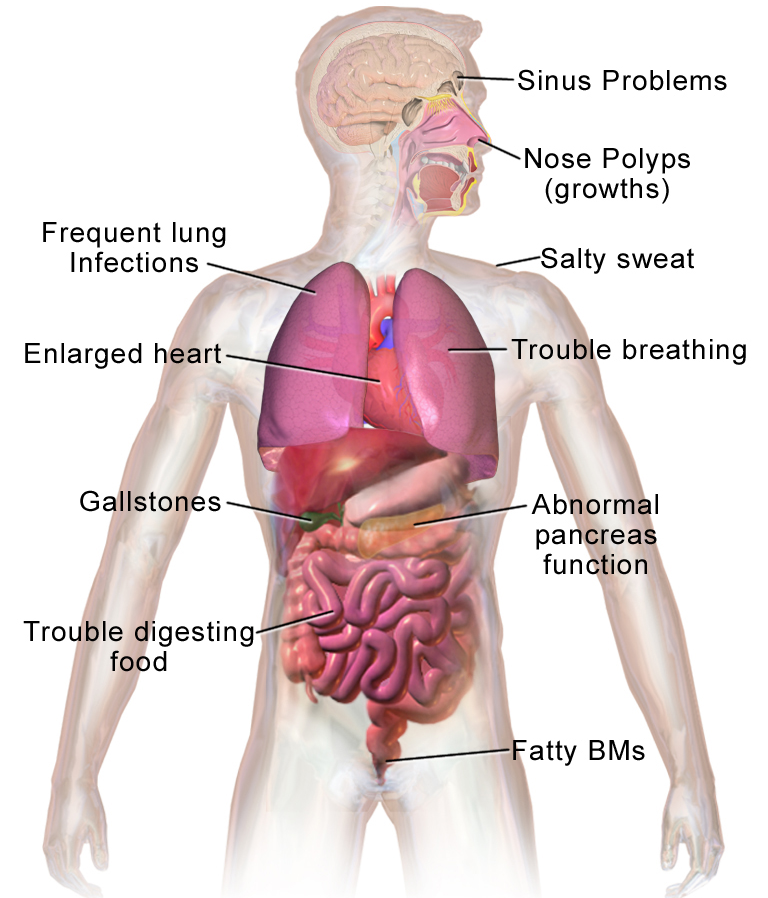
Mukowiscydoza (ang. cystic fibrosis, CF) to wrodzona choroba genetyczna, która wpływa na funkcjonowanie gruczołów wydzielających śluz i inne wydzieliny. W przebiegu choroby organizm wytwarza gęsty, lepki śluz, który zalega i utrudnia pracę wielu narządów — przede wszystkim płucach i układzie pokarmowym, ale też w innych częściach ciała. Choroba jest dziedziczona w sposób autosomalny recesywny: aby dziecko zachorowało, musi otrzymać wadliwy gen od obojga rodziców.
Galeria obrazów
4 Obrazy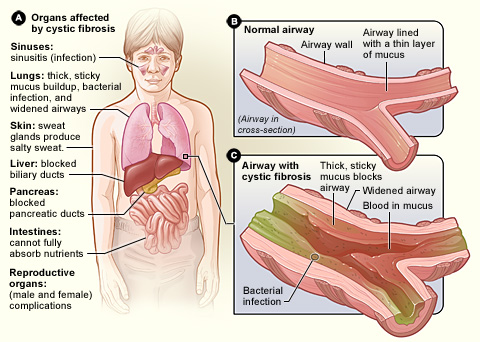

Przyczyny i genetyka
Mukowiscydozę wywołuje mutacja w genie CFTR, który koduje białko odpowiedzialne za transport jonów chloru i sodu przez błony komórkowe. Istnieje wiele różnych mutacji tego genu; najczęściej opisywana to mutacja ΔF508. Osoba, która otrzymała wadliwy gen tylko od jednego z rodziców, jest nosicielem (nie musi mieć objawów). Jeśli oboje rodzice są nosicielami tej samej wadliwej mutacji i oboje przekażą ją potomstwu, dziecko będzie chore. Nosicielstwo można wykryć badaniami genetycznymi.
Objawy
Objawy mukowiscydozy są zmienne — zależą od nasilenia choroby i rodzaju mutacji. Do najczęstszych należą:
- Objawy ze strony układu oddechowego: przewlekły kaszel, nawracające zapalenia płuc, zaleganie gęstego śluzu, duszność, chrypka, nawracające infekcje bakteryjne.
- Objawy ze strony układu pokarmowego: problemy z trawieniem i wchłanianiem (szczególnie tłuszczów), tłuste i obfite stolce, niewystarczający przyrost masy ciała, niedobory witamin rozpuszczalnych w tłuszczach (A, D, E, K).
- Niepłodność u mężczyzn: często wynikająca z wrodzonego braku nasieniowodów.
- Inne: słony pot (wysokie stężenie jonów w pocie), niedobór wzrostu u dzieci, polipy nosa, osteopenia/osteoporoza w przebiegu przewlekłym.
Ważne: osoba chora na mukowiscydozę sama w sobie nie „zaraża” osób zdrowych. Jednak pacjenci z CF mogą przenosić między sobą specyficzne drobnoustroje (np. Burkholderia cepacia), dlatego obowiązują zasady zapobiegania krzyżowym zakażeniom w środowisku opieki zdrowotnej i grupach pacjentów.
Rozpoznanie
- Badania przesiewowe noworodków: w wielu krajach wykonywane rutynowo; pozwalają na wczesne wykrycie choroby.
- Test potowy (badanie chlorków w pocie): klasyczne i nadal podstawowe badanie diagnostyczne — podwyższone stężenie chlorków potwierdza rozpoznanie u odpowiednio sklasyfikowanych pacjentów.
- Badania genetyczne: identyfikacja mutacji w genie CFTR potwierdza rozpoznanie i pomaga w doborze terapii (np. leków modyfikujących funkcję białka CFTR).
- Dodatkowe badania: testy czynnościowe płuc (spirometria), badania obrazowe (RTG, HRCT), ocena stanu odżywienia i funkcji trzustki.
Leczenie i opieka
Nie ma jednego uniwersalnego leku, który całkowicie wyleczy mukowiscydozę, ale dostępne są terapie, które znacznie poprawiają jakość i długość życia. Leczenie jest wielospecjalistyczne i dostosowane indywidualnie.
- Fizjoterapia oddechowa (techniki drenażu): codzienne techniki oczyszczania dróg oddechowych, ćwiczenia oddechowe, urządzenia ułatwiające usuwanie wydzieliny.
- Leki inhalowane: m.in. roztwory soli fizjologicznej o wyższym stężeniu (hypertoniczne), dornaza alfa (rozrzedzająca śluz), bronchodilatatory.
- Antybiotyki: do leczenia i zapobiegania zaostrzeniom zakażeń płucnych; stosowane doustnie, dożylnie lub drogą wziewną (np. tobramycyna).
- Leki modulujące CFTR: rewolucyjne leki, które poprawiają funkcję wadliwego białka CFTR u osób z niektórymi typami mutacji. Przykłady to: iwakaftor, lumakaftor/iwakaftor, tezaftor/iwakaftor i kombinacje z elexacaftorem. Nie wszystkie mutacje kwalifikują się do każdej terapii — decyzję podejmuje lekarz na podstawie genotypu.
- Wsparcie żywieniowe: dieta wysokoenergetyczna, suplementacja enzymów trzustkowych (jeśli występuje ich niewydolność), uzupełnianie witamin A, D, E i K.
- Opieka specjalistyczna: regularne kontrole w ośrodkach CF, szczepienia (zgodnie z zaleceniami), rehabilitacja i wsparcie psychologiczne.
- Transplantacja płuc: rozważana w ciężkich, zaawansowanych przypadkach niewydolności oddechowej jako ostateczność.
Zapobieganie i poradnictwo genetyczne
Dla par planujących potomstwo dostępne są badania przesiewowe nosicielstwa oraz poradnictwo genetyczne, które pozwalają ocenić ryzyko urodzenia dziecka chorego. W przypadku stwierdzonego nosicielstwa można rozważyć diagnostykę prenatalną lub badania przedimplantacyjne w procedurach IVF.
Rokowanie i badania nad leczeniem
Dzięki postępom w leczeniu pacjenci z mukowiscydozą żyją coraz dłużej i prowadzą pełniejsze życie. Rokowanie zależy od ciężkości choroby, częstości zaostrzeń i wczesnego wdrożenia opieki. Trwają badania nad nowymi terapiami, w tym lepszymi modulatorami CFTR, terapiami genowymi i innymi metodami zmniejszającymi obciążenie chorobą.
Podsumowanie: Mukowiscydoza to przewlekła choroba genetyczna wymagająca wielodyscyplinarnego podejścia — wczesne rozpoznanie, stała opieka specjalistyczna, odpowiednia farmakoterapia, fizjoterapia oddechowa i wsparcie żywieniowe znacząco poprawiają jakość życia i rokowanie pacjentów. Osoby z podejrzeniem nosicielstwa lub zdiagnozowaną chorobą powinny korzystać z poradnictwa genetycznego i opieki w wyspecjalizowanych ośrodkach.
Jeśli chcesz, mogę dodać informacje o konkretnych lekach modulujących CFTR, sposobach fizjoterapii oddechowej lub wskazówkach żywieniowych — napisz, która z tych sekcji Cię interesuje.
Co CF robi z ciałem
Mukowiscydoza wpływa na cały organizm. Ogólnie rzecz biorąc, organizm ma problemy z przemieszczaniem soli do tych części ciała, które jej potrzebują. Ponieważ organizm ma problemy z przemieszczaniem soli, gromadzi się ona w miejscach, w których nie powinna się znajdować, takich jak płuca, żołądek i jelita.
Płuca
W płucach, kiedy sól utknie, powoduje, że jest mniej wody, co sprawia, że śluz staje się bardzo gęsty. Oddychanie staje się bardzo trudne. Leczenie polega na podawaniu leków ułatwiających oddychanie, które dodają wody do płuc, dzięki czemu śluz staje się cieńszy i łatwiej go wykrztusić. Kiedy jest cieńszy i mniej śluzu, łatwiej jest oddychać.
Leczenie
Nie ma lekarstwa na mukowiscydozę. Nawet jeśli ludzie mogą robić rzeczy, aby pozostać zdrowym. Zdrowe nawyki utrzymać osobę od coraz bardziej chory. Ludzie mogą zachować czystość. Ludzie mogą trzymać się z dala od zarazków. Mogą pić wodę, aby pomóc śluzu odejść. Przyjmowanie enzymów pomaga w trawieniu pokarmów, jeśli w żołądku jest śluz.
Ćwiczenie oczyszcza śluz. Buduje silne mięśnie i kości oraz wzmacnia płuca. Przyjmowanie witamin pomaga organizmowi zwalczyć infekcję. Pomaga także ciału rosnąć i dobrze funkcjonować.
- Wziewne antybiotyki są stosowane w celu powstrzymania bakterii przed rozwojem w gęstym śluzie.
- Wdychana słona woda pomaga utrzymać nawilżenie płuc
Badanie w kierunku mukowiscydozy
- Test chlorkowy potu - bada poziom soli w pocie danej osoby.
- Test genetyczny - jest stosowany, jeśli test potowy jest pozytywny, aby sprawdzić, czy mają oba geny.
65 róż
"65 róż" to sposób, w jaki niektóre dzieci odnoszą się do swojego stanu, ponieważ mukowiscydoza jest trudna do wypowiedzenia przez małe dziecko. 65 róż" jest również zastrzeżonym znakiem towarowym Fundacji Mukowiscydozy, aby pomóc kontrolować jego użycie. Jest to bardzo pomocny sposób na zrozumienie przez małe dzieci. Wypowiadane na głos brzmi podobnie do mukowiscydozy.
Pytania i odpowiedzi
P: Co to jest mukowiscydoza?
Mukowiscydoza to choroba powodująca wytwarzanie przez organizm gęstego, lepkiego śluzu, który może gromadzić się w płucach, układzie pokarmowym i innych częściach ciała.
P: Jak powstaje mukowiscydoza?
Mukowiscydoza jest spowodowana odziedziczeniem genu mukowiscydozy od obojga rodziców.
P: Czy osoba posiadająca tylko jeden gen mukowiscydozy może przekazać chorobę swojemu dziecku?
O: Osoba posiadająca tylko jeden gen mukowiscydozy może sama nie być chora na tę chorobę, ale nadal może przekazać ten gen swojemu dziecku.
P: Czy mukowiscydoza jest zaraźliwa?
Mukowiscydoza nie jest zaraźliwa i nie może być przenoszona z jednej osoby na drugą.
P: Czy istnieje lekarstwo na mukowiscydozę?
O: Nie, obecnie nie ma lekarstwa na mukowiscydozę, ale istnieje wiele leków, które mogą pomóc w leczeniu tej choroby.
P: Jakie części ciała mogą być dotknięte mukowiscydozą?
Mukowiscydoza może wpływać na płuca, układ trawienny i inne części ciała.
P: Jak osoby z mukowiscydozą mogą zachować zdrowie?
O: Osoby z mukowiscydozą mogą zachować zdrowie, przyjmując leki, monitorując swój stan i prowadząc zdrowy tryb życia.
Powiązane artykuły
Autor
AlegsaOnline.com Mukowiscydoza — definicja, przyczyny, objawy i leczenie Leandro Alegsa
URL: https://pl.alegsaonline.com/art/24952