Reakcje elektrocykliczne w chemii organicznej — definicja, mechanizm, przykłady
Reakcje elektrocykliczne w chemii organicznej — definicja, mechanizm i przykłady. Poznaj reguły Woodward–Hoffmann, elektrocyklizację, stereoselektywność i praktyczne zastosowania.
W chemii organicznej reakcja elektrocykliczna jest szczególnym rodzajem peryklicznej reakcji przegrupowania. Reakcja jest elektrocykliczna, gdy w jej wyniku jedno wiązanie pi ulega przemianie w wiązanie sigma (zamknięcie pierścienia, elektrocyklizacja) lub gdy jedno wiązanie sigma ulega przekształceniu w wiązanie pi (otwieranie pierścienia). Reakcje te przebiegają przez zorganizowane, cykliczne przejście orbitalne i wykazują kilka charakterystycznych cech:
- reakcje elektrocykliczne mogą być napędzane przez światło (fotoindukowane) lub przez ciepło (termiczne) — w zależności od sposobu wzbudzenia elektrony zajmują inne orbitale;
- tryb reakcji (czyli czy przebiega konrotacyjnie czy dysrotacyjnie) jest określony liczbą elektronów π w układzie koniugowanym — stąd zależność od reguł symetrii orbitalnej;
- reakcja elektrocykliczna może zamknąć pierścień (elektrocyklizacja) lub otworzyć pierścień (np. otwarcie cyklobutenu do dienu);
- stereospecyficzność produktu jest określana przez sposób tworzenia się przejściowego — konrotacyjny lub dysrotacyjny — zgodnie z zasadami Woodward–Hoffmanna.
Galeria obrazów
6 Obrazy



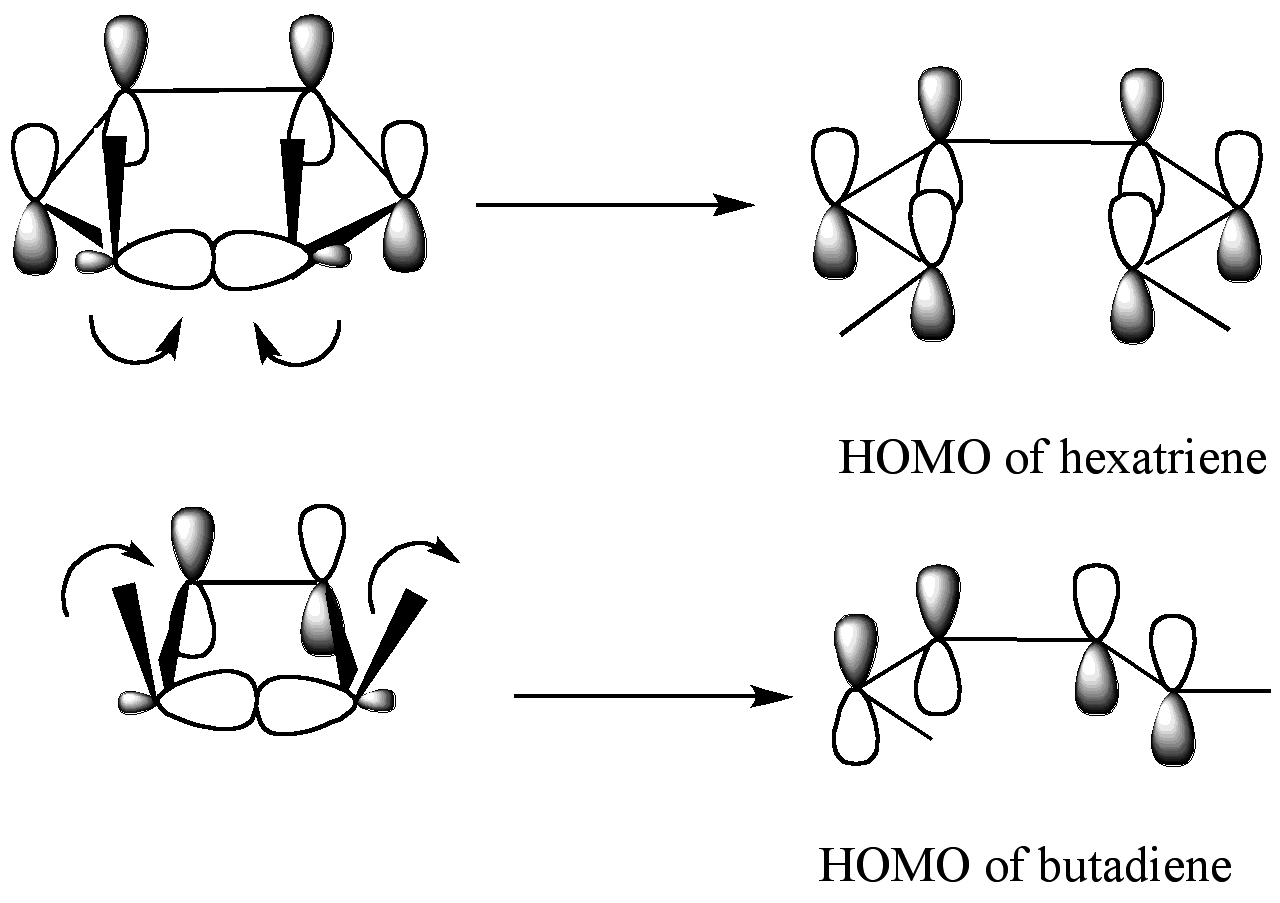

Podstawy teoretyczne: reguły Woodwarda–Hoffmanna i orbitale brzegowe
Kluczowym pojęciem wyjaśniającym wybór mechanizmu jest konserwacja symetrii orbitalnej. Zasady Woodwarda–Hoffmanna przewidują, które elektrocyklizacje są dozwolone (tj. przebiegają łatwo) w warunkach termicznych, a które są dozwolone w warunkach fotochemicznych. W praktyce stosuje się prostą regułę elektryczną opartą na liczbie elektronów π:
- Systemy 4n+2 elektronów π (np. 2π, 6π): termicznie preferują przebieg dysrotacyjny, fotochemicznie — konrotacyjny.
- Systemy 4n elektronów π (np. 4π, 8π): termicznie preferują przebieg konrotacyjny, fotochemicznie — dysrotacyjny.
Wyjaśnienie orbitalne (metoda frontier-orbital) polega na porównaniu symetrii najwyżej obsadzonego orbitalu molekularnego (HOMO) reaktanta z symetrią orbitali produktu. Podczas rozrywania wiązania sigma i tworzenia pary orbitali π nowo powstałe p-orbitaly muszą być kompatybilne symetrycznie z HOMO produktu, aby przebieg był sprzyjany energetycznie.
Konrotacja i dysrotacja — co to oznacza stereochemicznie?
W elektrocyklizacji opisujemy dwa typy ruchu końców łańcucha prowadzącego do zamknięcia pierścienia:
- konrotacja (conrotatory) — oba końce łańcucha obracają się w tym samym kierunku (np. oba w prawo lub oba w lewo);
- dysrotacja (disrotatory) — końce łańcucha obracają się w przeciwnych kierunkach.
Ten wybór rotacji determinuje końcową konfigurację podstawników przy nowo powstałym wiązaniu sigma — od tego zależy, czy otrzymamy izomery cis/trans lub pary enancjomeryczne.
Torquoselektywność (momentoselektywność)
Momentoselektywność (z ang. torquoselectivity, czasem tłumaczona jako selektywność skrętu) odnosi się do kierunku, w którym obracają się podstawniki podczas procesu konrotacji lub dysrotacji. Nawet jeśli mechanizm dopuszcza obrót w obu kierunkach, efekty elektronowe i steryczne podstawników mogą ograniczać preferowany kierunek obrotu i prowadzić do przewagi jednego stereoisomeru nad drugim. Przy pełnej selektywności otrzymujemy niemal wyłącznie jeden stereoizomer; przy częściowej — mieszaninę z przewagą jednego z nich.
Przykłady i ilustracje
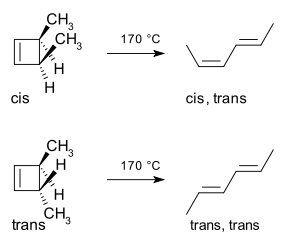
Klasycznym przykładem jest termiczne otwarcie pierścienia 3,4-dimetylocyklobutenu. Izomer cis daje wyłącznie cis,trans-2,4-heksadien, podczas gdy izomer trans prowadzi do trans,trans-2,4-heksadien. Ten stereospecyficzny wynik wynika z mechanizmu otwarcia pierścienia (konrotacyjnego lub dysrotacyjnego) i zgodności z symetrią orbitalną:
Metoda orbitali brzegowych wyjaśnia szczegóły: wiązanie sigma w reagencie otwiera się tak, że powstałe p-orbity mają tę samą symetrię co HOMO produktu (butadien). W omawianym przypadku otwarcie pierścienia zachodzi w sposób rotacyjny, który daje odpowiednie znaki fazowe orbitali po obu stronach przerwanej wiązki, co jest zgodne z formacją dozwolonego produktu:
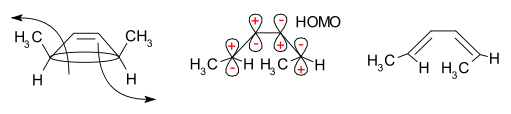
Nazarowska cyklizacja — specjalny przypadek
Nazarowska reakcja cyklizacyjna to przykład elektrocyklizacji zamykającej pierścień. Nazarow (Iwan N. Nazarow) odkrył, że diwinyloketony mogą ulegać elektrocyklizacji prowadzącej do cyklopentenonów — proces wykorzystywany często w syntezie złożonych systemów cyklicznych. Mechanizm typowo przebiega przez uwodornienie i powstanie karbokationu, który następnie zamyka się do cyklopentenonu; reakcja ta ma wielkie znaczenie w syntezach naturalnych i przemysłowych.
Inne przykłady i zastosowania
- 6π-elektrocyklizacja all-triene do cykloheksadienu — istotna w tworzeniu sześcioczłonowych układów aromatycznych i bicyklicznych struktur;
- fotochemiczne elektrocyklizacje stosowane do tworzenia cyklicznych struktur w syntezach asymetrycznych;
- wykorzystanie torquoselektywności i kataliz chemicznych do uzyskiwania wysokiej selektywności enancjomerycznej.
Stereospecyficzność, dowody eksperymentalne i praktyczne uwagi
Chemicy interesują się reakcjami elektrocyklicznymi, ponieważ ich stereochemia i mechanika potwierdzają przewidywania teoretyczne dotyczące zachowania symetrii orbitali molekularnych oraz zasad Woodwarda–Hoffmanna. Doświadczenia z izomerami geometrycznymi, badania kinetyczne oraz spektroskopia (np. NMR) pozwalają śledzić, czy reakcja przebiega konrotacyjnie czy dysrotacyjnie i jakimi produktami kończy się proces. W praktyce wpływ na wynik mają temperatura, światło, podstawnikowanie substratów oraz obecność katalizatorów lub czynników jonizujących.
Podsumowanie
Reakcje elektrocykliczne to ważna klasa reakcji pericyklicznych, łącząca wymiar teoretyczny (symetria orbitali, reguły Woodwarda–Hoffmanna) z praktyką syntez organicznych. Znajomość zasad konrotacji/dysrotacji, zależności od liczby elektronów π oraz efektów sterycznych i elektronowych podstawników pozwala przewidywać i kontrolować stereochemię otrzymywanych produktów.
Zasady Woodwarda-Hoffmana
Zasady Woodwarda-Hoffmanna dotyczą zachowania symetrii orbitalnej w reakcjach elektrocyklicznych.

Schematy korelacji łączą orbitę molekularną reaktora z orbitami produktu o tej samej symetrii. Wykresy korelacji mogą być rysowane dla tych dwóch procesów.
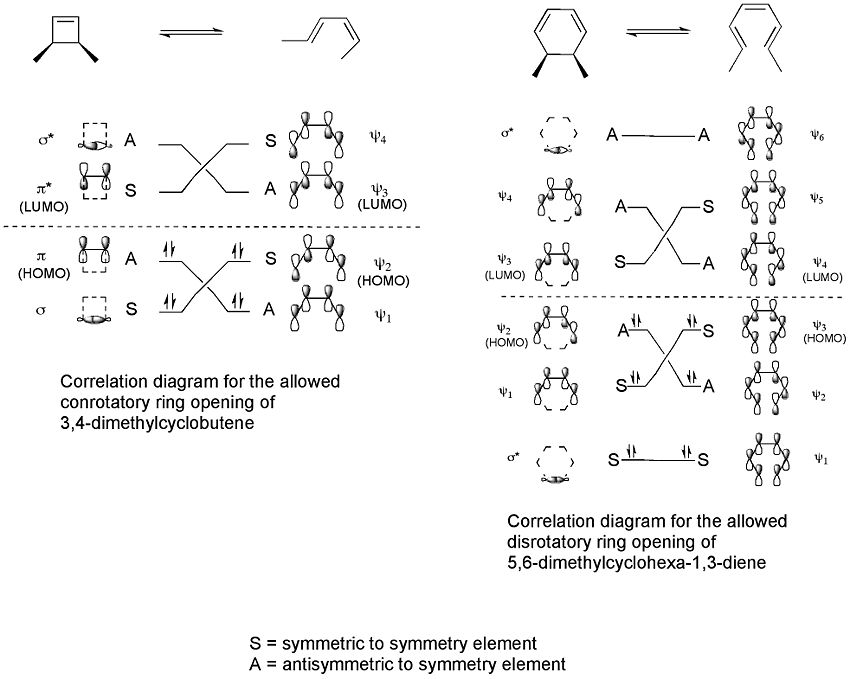
Te schematy korelacji wskazują, że tylko rotacyjny otwór pierścienia 3,4-dimetylocyklobutenu jest "symetryczny", podczas gdy tylko rotacyjny otwór pierścienia 5,6-dimetylocykloheksa-1,3-dienu jest "symetryczny". Wynika to z faktu, że tylko w tych przypadkach w stanie przejściowym występuje maksymalne nakładanie się na siebie orbitalne. Również uformowany produkt znajdowałby się raczej w stanie uziemienia niż pobudzenia.
Graniczna teoria molekularna orbitalna
Frontier Molecular Orbital Theory przewiduje, że wiązanie sigma w pierścieniu otworzy się w taki sposób, że powstałe p-orbity będą miały taką samą symetrię jak HOMO produktu.
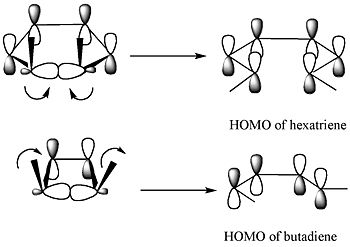
Powyższy wykres przedstawia dwa przykłady. Dla 5,6-dimetylocykloheksa-1,3-dienu (górny rząd wykresu), tylko tryb rotacyjny spowoduje, że p-orbity będą miały taką samą symetrię jak HOMO heksatrienu. Dwa p-orbity obracają się w przeciwnych kierunkach. Dla 3,4-dimetylocyklobutenu (dolny rząd wykresu), tylko tryb rotacyjny dawałby p-orbity o tej samej symetrii co HOMO butadienu. P-oribtale obracają się w tym samym kierunku.
Elektrocyklizacje stanu podnieconego
Światło może przemieszczać elektron do stanu pobudzenia, który zajmuje wyższą orbitę. Elektron wzbudzony będzie zajmował LUMO, który ma wyższy poziom energii niż stara orbita elektronu. Jeśli światło otworzy pierścień 3,4-dimetylocyklobutenu, wynikowa elektrocyklizacja nastąpi w trybie rotacyjnym, a nie w trybie rotacyjnym. Wykres korelacji dla dopuszczalnej reakcji otwarcia pierścienia stanu wzbudzonego pokazuje dlaczego:
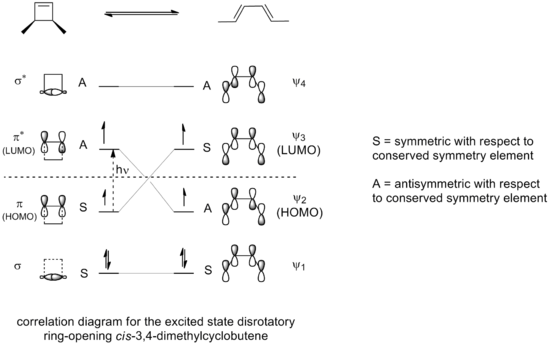
Tylko tryb rotacyjny, w którym symetria o płaszczyźnie odbicia jest utrzymywana przez cały czas trwania reakcji, skutkowałby maksymalnym nakładaniem się na siebie orbit w stanie przejściowym. Ponadto, po raz kolejny, spowodowałoby to powstanie produktu, który jest w stanie wzbudzonym o porównywalnej stabilności do stanu wzbudzonego związku reaktywnego.
Reakcje elektrocykliczne w układach biologicznych
Reakcje elektrocykliczne występują często w przyrodzie. Jedną z najczęstszych takich reakcji w przyrodzie jest biosynteza witaminy D3.
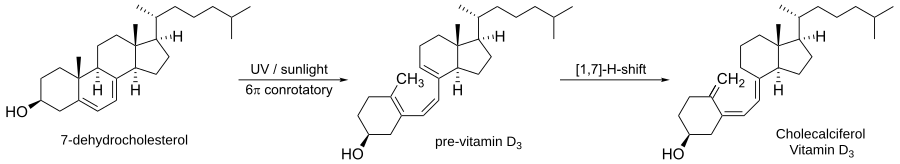
Pierwszy krok polega na lekkim otwarciu pierścienia 7-dehydrocholesterolu w celu utworzenia prewitaminy D3. Jest to fotochemicznie indukowana elektrocykliczna reakcja rotacyjna. Drugim krokiem jest [1,7]- przesunięcie bezwodnika, aby utworzyć witaminę D3.
Innym przykładem jest proponowana biosynteza aranotyny, oksepiny występującej w przyrodzie i związanych z nią związków.
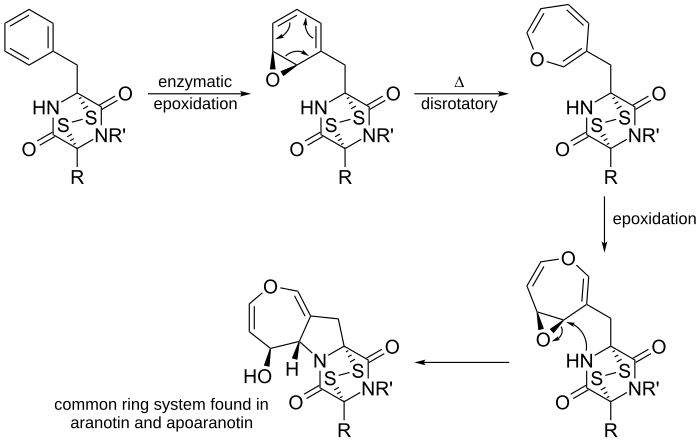
Fenyloalanina jest używana do produkcji diketopiperazyny (nie pokazano). Następnie enzymy epoksydują diketopiperazynę w celu utworzenia tlenku areny. Poddaje się ją reakcji elektrocyklizacji otwierającej pierścień rotacyjny o 6π w celu wytworzenia niecyklizowanej oksepiny. Po drugiej epoksydacji pierścienia, pobliski nukleofilowy azot atakuje elektrofilowy węgiel, tworząc pięciopunktowy pierścień. Powstały w ten sposób układ pierścieniowy jest wspólnym układem pierścieni znalezionym w aranotynie i związanych z nią związkach.
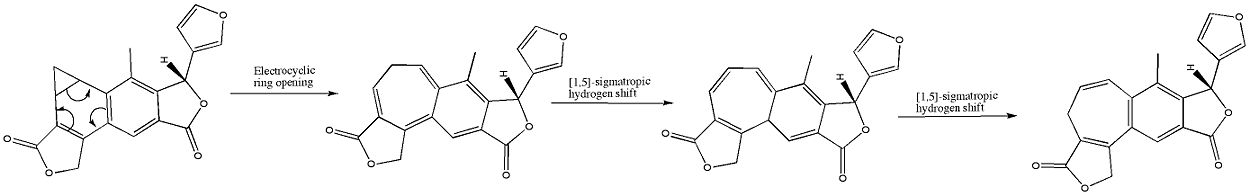
Diterpenoid benzonorkaradienu (A) został przekształcony w diterpenoid benzocykloheptatrienu (B) poprzez gotowanie roztworu chlorku metylenu. Transformację tę można uznać za rotacyjną elektrocykliczną reakcję, po której nastąpiły dwie ponadtwardowe, 1,5-metropowe zmiany wodoru, jak pokazano poniżej:
Zakres stosowania
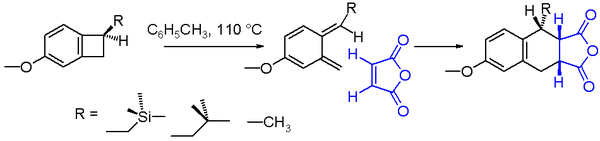
Przykładem reakcji elektrocyklicznej jest rotacyjne termiczne otwarcie pierścienia benzocyklobutanu. Produktem reakcji jest bardzo niestabilny ortochinodimetan. Cząsteczka ta może być uwięziona w endo-dodatku o silnym dienofilu, takim jak bezwodnik maleinowy do adduktu Diels-Alder. Stwierdzono, że wydajność chemiczna otworu pierścieniowego benzocyklobutanu przedstawionego na schemacie 2 zależy od rodzaju podstawnika R. W przypadku rozpuszczalnika reakcyjnego takiego jak toluen i temperatury reakcji 110 °C wydajność wzrasta, przechodząc od metylu do izobutylo-metylu do trimetylosililometylu. Wzrost szybkości reakcji dla związku trimetylosilililowego można wytłumaczyć hiperkoniugacją krzemu, ponieważ wiązanie βC-Si osłabia wiązanie cyklobutanu C-C poprzez dawkowanie elektronów.
Odkryto biomimetyczną elektrocykliczną reakcję kaskadową w odniesieniu do izolacji i syntezy niektórych kwasów endiandrycznych:

Pytania i odpowiedzi
P: Co to jest reakcja elektrocykliczna?
O: Reakcja elektrocykliczna jest rodzajem reakcji rearanżacji perycyklicznej, w której wyniku jedno wiązanie pi staje się jednym wiązaniem sigma lub jedno wiązanie sigma staje się wiązaniem pi.
P: Jak przebiegają reakcje elektrocykliczne?
O: Reakcje elektrocykliczne są napędzane przez światło (fotoindukcja) lub ciepło (termiczne).
P: Jak liczba elektronów pi wpływa na reakcję elektrocykliczną?
O: Liczba elektronów pi wpływa na tryb reakcji w reakcji elektrocyklicznej.
Q: Co się dzieje podczas procesu elektrolizy?
O: Podczas procesu elektrolizy może dojść do zamknięcia pierścienia.
P: Co decyduje o stereospecyficzności w reakcji elektrocyklicznej?
O: Stereotypowość w reakcji elektrocyklicznej jest określana przez konrotacyjne lub dysrotacyjne tworzenie stanu przejściowego, zgodnie z regułami Woodwarda-Hoffmanna.
P: Czym jest torquoselektywność w odniesieniu do reakcji elektrocyklicznej?
O: Torkooselektywność odnosi się do kierunku, w którym obracają się podstawniki podczas reakcji elektrocyklicznej, w wyniku której mogą powstać enancjomeryczne produkty, jeżeli przebiega ona w procesie konrotacyjnym, i nadmiar enancjomerów, jeżeli przebiega ona w procesie torkooselektywnym.
P:Jaki przykład ilustruje, jak metoda frontier-orbital wyjaśnia to działanie?
O: Reakcja termicznego otwarcia pierścienia 3,4-dimetylocyklobutenu jest przykładem ilustrującym, jak metoda frontier-orbital wyjaśnia, jak to działa. Wiązanie sigma otworzy się w taki sposób, że powstałe p-orbitale będą miały taką samą symetrię jak najwyżej położony orbital molekularny (HOMO) produktu (butadienu). Dzieje się tak tylko przy konrotacyjnym otwarciu pierścienia, które powoduje przeciwne znaki dla dwóch płatów na przerwanych końcach pierścienia, podczas gdy dysrotacyjne tworzyłoby anty wiązanie.
Powiązane artykuły
Autor
AlegsaOnline.com Reakcje elektrocykliczne w chemii organicznej — definicja, mechanizm, przykłady Leandro Alegsa
URL: https://pl.alegsaonline.com/art/30716
Źródła
- iupac.org : "electrocyclic reaction"
- dx.doi.org : 10.1016/S0040-4039(01)83997-6
- dx.doi.org : 10.1021/ar50001a003
- dx.doi.org : 10.1021/cr0406110
- dr.doi.org : Abstract
- dx.doi.org : 10.1021/ja00384a080
- dx.doi.org : 10.1021/jo802351b