Orbital molekularny — definicja, teoria i zastosowania w chemii kwantowej
Orbital molekularny: przystępne wyjaśnienie definicji, teorii i zastosowań w chemii kwantowej — modele, obliczenia i praktyczne przykłady dla badaczy i studentów.
W chemii, orbital molekularny (lub MO) wyjaśnia, co dzieje się z elektronami, gdy atomy łączą się w cząsteczce. MO jest matematyczną funkcją, która opisuje falowe zachowanie elektronu w cząsteczce. Chemicy używają takich funkcji do przewidywania lub wyjaśniania właściwości chemicznych i fizycznych. Na przykład, funkcje te mogą określić prawdopodobieństwo znalezienia elektronu w dowolnym określonym obszarze.
Chemicy zazwyczaj budują modele matematyczne orbit molekularnych poprzez łączenie orbit atomowych. Można również wykorzystać orbitały hybrydowe z każdego atomu cząsteczki, lub inne orbitały molekularne z grup atomów. Na tych funkcjach mogą pracować komputery. Orbitale molekularne pozwalają chemikom na zastosowanie mechaniki kwantowej do badania cząsteczek. MOs odpowiadają na pytania o to, jak atomy w cząsteczkach łączą się ze sobą. Różne zaokrąglone kształty na schemacie orbitalnym wskazują, gdzie najprawdopodobniej znajdowałyby się elektrony w atomie.
Galeria obrazów
5 Obrazy
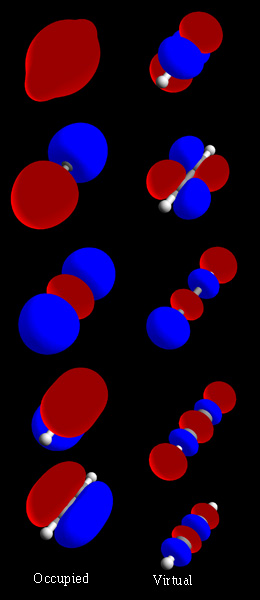



Jak powstają orbitale molekularne — metoda LCAO
Najprostszy i najczęściej stosowany sposób konstrukcji orbitali molekularnych to metoda LCAO (Linear Combination of Atomic Orbitals) — liniowa kombinacja orbit atomowych. W tej koncepcji orbity atomowe poszczególnych atomów są ważone i sumowane, tworząc nowe funkcje rozciągające się na całą cząsteczkę. Sumy te dają dwa podstawowe typy orbitali:
- Orbital wiążący — powstaje przez konstruktywną interferencję falową (sumowanie faz zgodnych); ma niższą energię niż odpowiadające orbitale atomowe i prowadzi do stabilizacji wiązania.
- Orbital antywiążący — powstaje przez destruktywną interferencję (sumowanie faz przeciwnych); ma wyższą energię niż orbitale atomowe i osłabia wiązanie.
- Orbital niesparowany (wolna para / nonbonding) — orbita, która pozostaje w przybliżeniu na jednym atomie i nie przyczynia się istotnie do stabilizacji lub destabilizacji wiązania.
Symetria i reguły łączenia orbitali
Przy łączeniu orbitali ważna jest zgodność symetrii i energii. Orbital atomowy o pewnej symetrii może tworzyć z innym orbitale tylko wtedy, gdy mają zgodne właściwości symetryczne względem osi i płaszczyzn w cząsteczce. Różnice energii między orbitami atomowymi wpływają na stopień mieszania — im bliżej energetycznie, tym większe mieszanie i bardziej rozciągły orbital molekularny.
Diagramy orbitalne, obsada, HOMO i LUMO
Po zbudowaniu orbitali przypisuje się do nich obsadę elektronową zgodnie z zasadą zakazu Pauliego i regułą Hund'a. Kluczowe pojęcia to:
- HOMO (Highest Occupied Molecular Orbital) — najwyższy zajęty orbital molekularny; często determinuje zdolność do oddawania elektronu (kwasowość, reakcje utleniania).
- LUMO (Lowest Unoccupied Molecular Orbital) — najniższy niezajęty orbital molekularny; jest akceptorem elektronu w reakcjach — określa podatność na przyjmowanie elektronu (reaktywność elektrofilowa).
- Porządek wiązania — obliczany jako (liczba elektronów w orbitalach wiążących − liczba elektronów w orbitalach antywiążących) / 2; daje przybliżone pojęcie o sile wiązania.
Przykłady prostych cząsteczek
W najprostszych przypadkach, jak H2, dwa atomowe orbitale 1s łączą się tworząc jeden orbital wiążący σ(1s) i jeden antywiążący σ*(1s); wypełnienie orbitalu wiążącego dwoma elektronami daje trwałe wiązanie. W cząsteczce O2 teoria MO tłumaczy jej paramagnetyzm — dwa niesparowane elektrony zajmują orbitale π* antywiążące, co prowadzi do właściwości magnetycznych, których nie wytłumaczyłby prosty model wiązań kowalencyjnych.
Zastosowania w chemii i materiałach
Orbitalne opisywanie elektronów jest fundamentem wielu dziedzin chemii kwantowej i technologii:
- Wyjaśnianie i przewidywanie geometrii cząsteczek, długości i siły wiązań.
- Badanie reaktywności chemicznej — analiza HOMO/LUMO pozwala przewidywać miejsca ataku nukleofilowego i elektrofilowego.
- Spektroskopia — przejścia elektronowe między MO odpowiadają liniom w UV‑Vis, fotoemisji itp.
- Projektowanie materiałów przewodzących i półprzewodników — pasma energetyczne w ciałach stałych są rozszerzeniem pojęcia MO dla dużych układów.
- Kataliza i teoria reaktywności — orbity przejściowe i hybrydowe wyjaśniają mechanizmy katalityczne i wybiórczość reakcji.
Metody obliczeniowe i wizualizacja
Do obliczeń używa się wielu metod: przybliżonych (Hückel, ucząc się z prostych modeli) oraz bardziej zaawansowanych z zakresu mechaniki kwantowej, takich jak Hartree–Fock, metody post‑Hartree–Fock (MP2, CC) i funkcjonały gęstości (DFT). Wynikiem obliczeń są funkcje falowe i rozkłady gęstości elektronowej, które można wizualizować jako izopowierzchnie pokazujące kształt MW. Na tych wizualizacjach łatwiej jest zrozumieć rozkład ładunku, polaryzację i charakter wiązań.
Orbitały hybrydowe i lokalizacja
W wielu cząsteczkach wygodniej jest myśleć o orbitałach lokalnych lub hybrydowych (np. sp, sp2, sp3) — to transformacje liniowe MO, które ułatwiają interpretację geometrii i chemii wiązań (np. kątów i kierunkowości wiązań w związkach organicznych). Lokalizowane MO (np. orbitale Boys'a) pozwalają na oddzielenie wiązań kowalencyjnych i wolnych par w wygodny sposób do analizy chemicznej.
Ograniczenia i przybliżenia
Model MO jest potężny, ale opiera się na przybliżeniach: w praktyce używa się skończonych baz funkcji, zaniedbuje niektóre korelacje elektronowe lub stosuje przybliżone funkcjonały DFT. Dlatego wyniki powinny być interpretowane z uwzględnieniem błędów metodycznych oraz porównywane z danymi eksperymentalnymi tam, gdzie to możliwe.
Podsumowanie
Orbitale molekularne to centralne narzędzie chemii kwantowej: łącząc idee falowe z zasadami symetrii i regułami obsady, pozwalają chemikom przewidywać i wyjaśniać strukturę, właściwości i reaktywność cząsteczek. Dzięki komputerowym metodom obliczeniowym MO stały się praktycznym instrumentem w projektowaniu leków, katalizatorów, materiałów oraz w zrozumieniu procesów fotochemicznych i elektronowych.

Historia
Słowo "orbital" zostało po raz pierwszy użyte w języku angielskim przez Roberta S. Mullikena. O MO pisał wcześniej niemiecki fizyk Erwin Schrödinger. Schrödinger nazwał je Eigenfunktion.
Fizyk Max Born opisał teorię stojącą za orbitami molekularnymi w 1926 roku. Dziś jest ona znana jako reguła Borna i stanowi część kopenhaskiej interpretacji mechaniki kwantowej. Początkowo zaproponowana teoria ta nie zgadzała się z modelem atomu Nielsa Bohra. Model Bohra opisywał elektrony jako "orbitujące" wokół jądra, ponieważ poruszały się one w kółko. Jednakże, model Borna w końcu zyskał popularne poparcie, ponieważ był w stanie opisać położenie elektronów w cząsteczkach i wyjaśnić szereg wcześniej niewytłumaczalnych reakcji chemicznych.
Przegląd
Orbity atomowe przewidują położenie elektronu w atomie. Orbity molekularne powstają, gdy orbity atomowe są skupione razem. Orbita molekularna może dać informację o konfiguracji elektronu w cząsteczce. Konfiguracja elektronów jest najbardziej prawdopodobnym położeniem, a energia jednego (lub jednej) pary elektronów. Najczęściej MO jest reprezentowany jako liniowa kombinacja orbit atomowych (metoda LCAO-MO), szczególnie w przybliżeniu. Oznacza to, że chemicy zakładaj±, że prawdopodobieństwo istnienia elektronu w dowolnym punkcie cz±steczki jest sum± prawdopodobieństw istnienia tam elektronu w oparciu o poszczególne orbity atomowe. LCAO-MO jest prostym modelem wiązania w molekułach i jest ważny dla badania orbitalnej teorii molekularnej.
Chemicy teoretyczni używają komputerów do obliczania MO różnych molekuł (zarówno rzeczywistych, jak i wyimaginowanych). Komputer może narysować wykresy "chmury", aby pokazać jak prawdopodobny będzie elektron w dowolnym regionie. Komputer może również podać informacje o fizycznych właściwościach cząsteczki. Mogą również powiedzieć, ile energii potrzeba do utworzenia cząsteczki. To pomaga chemikom powiedzieć, czy niektóre małe molekuły mogą być połączone w większe molekuły.
Większość współczesnych sposobów wykonywania chemii obliczeniowej zaczyna się od obliczania MO systemu. Pole elektryczne każdego MO jest generowane przez jądra wszystkich atomów i jakiś średni rozkład pozostałych elektronów.
Analogia
Zrozumienie MOs jest jak zadanie poznania, gdzie każdy pracownik znajduje się w dużym sklepie z artykułami wyposażenia wnętrz (bez zaglądania do środka sklepu). Analityk zna liczbę pracowników pracujących w sklepie i w dziale każdego pracownika. Wie również, że pracownicy nie nadepną na siebie, a pracownicy staną w przejściu, a nie na półkach sklepowych. Pracownicy opuszczają swój własny dział, aby pomóc klientom zlokalizować towary w innych działach lub sprawdzić zapasy. Analityk podający lokalizację wszystkich pracowników w sklepie w wybranym momencie bez zaglądania do środka jest jak chemik obliczający MO molekuły. Tak jak MOs nie mogą podać dokładnej lokalizacji każdego elektronu, tak dokładne położenie każdego pracownika nie jest znane. MO posiadający płaszczyznę węzłową jest jak wniosek, że pracownicy chodzą po korytarzach, a nie po półkach. Chociaż elektrony pochodzą z konkretnego atomu, to jednak elektron wypełnia MO bez względu na swój atom źródłowy. To tak, jakby pracownik opuszczał swój dział, by w ciągu dnia chodzić gdzie indziej w sklepie. Tak więc, MO jest niekompletnym opisem elektronu, tak jak obliczenia analityka na temat niewidzialnego sklepu są niekompletnym zgadywaniem na temat lokalizacji pracowników.

Tworzenie orbit molekularnych
Chemicy teoretyczni wymyślili zasady obliczania MO. Reguły te pochodzą ze zrozumienia mechaniki kwantowej. Mechanika kwantowa pomaga chemikom wykorzystać to, co fizycy powiedzieli o elektronach, aby ustalić, jak zachowują się one w cząsteczkach. Orbity molekularne powstają w wyniku "dozwolonych" oddziaływań pomiędzy orbitami atomowymi. (Oddziaływania te są "dozwolone", jeżeli symetrie (określone na podstawie teorii grupowej) orbit atomowych są ze sobą kompatybilne). Chemicy badają oddziaływania z orbitami atomowymi. Oddziaływania te pochodzą z nakładania się (miara tego, jak dobrze dwie orbity konstruktywnie oddziałują na siebie) pomiędzy dwoma orbitami atomowymi. Nakładanie się jest ważne, jeżeli orbity atomowe są blisko siebie pod względem energetycznym. W końcu, liczba MO w cząsteczce musi być równa liczbie orbit atomowych w atomach, które są połączone, aby utworzyć cząsteczkę.
Podejście jakościowe
Chemicy muszą zrozumieć geometrię MOs, aby omówić strukturę molekularną. Metoda LCMO (Linear combination of atomic orbitals molecular orbital) daje przybliżony, ale dobry opis MOs. W tej metodzie, orbity molekularne są wyrażone jako liniowe kombinacje wszystkich orbit atomowych każdego atomu w cząsteczce.
Liniowe kombinacje orbit atomowych (LCAO)
Orbitale molekularne zostały po raz pierwszy wprowadzone przez Friedricha Hunda i Roberta S. Mullikena w 1927 i 1928 roku.
Liniowe połączenie orbit atomowych lub przybliżenie "LCAO" dla orbit molekularnych zostało wprowadzone w 1929 roku przez Sir Johna Lennarda-Jonesa. Jego przełomowy artykuł pokazał, jak wyprowadzić elektroniczną strukturę cząsteczek fluoru i tlenu z zasad kwantowych. To jakościowe podejście do molekularnej teorii orbitalnej jest częścią początków współczesnej chemii kwantowej.
Liniowe kombinacje orbit atomowych (LCAO) mogą być używane do odgadywania orbit molekularnych, które powstają, gdy atomy cząsteczki łączą się ze sobą. Podobnie jak w przypadku orbity atomowej, równanie Schrodingera, które opisuje zachowanie się elektronu, może być skonstruowane również dla orbity molekularnej. Liniowe kombinacje orbit atomowych, (sumy i różnice fali atomowej) dostarczają przybliżonych rozwiązań dla molekularnych równań Schrodingera. Dla prostych molekuł okrzemkowych, fale, które otrzymujemy, są reprezentowane matematycznie przez równania
Ψ = caψa + cbψb
oraz
Ψ* = caψa - cbψb
gdzie Ψ i Ψ* są falami molekularnymi dla orbit molekularnych wiążących i przeciwwiążących, odpowiednio ψa i ψb są falami atomowymi z atomów a i b, odpowiednio, a ca i cb są współczynnikami regulowanymi. Współczynniki te mogą być dodatnie lub ujemne, w zależności od energii i symetrii poszczególnych orbit atomowych. Gdy dwa atomy zbliżają się do siebie, ich orbity atomowe nakładają się na siebie, tworząc obszary o dużej gęstości elektronów. Tak więc, molekularne orbity są tworzone pomiędzy dwoma atomami. Atomy są trzymane razem przez elektrostatyczne przyciąganie pomiędzy dodatnio naładowanymi jądrami i ujemnie naładowanymi elektronami zajmującymi wiążące orbity molekularne.
Wiązania, Antibonding i Nonbonding MOs
Kiedy orbity atomowe oddziaływują na siebie, powstająca orbita molekularna może mieć trzy rodzaje: wiążącą, antywiążącą lub nie wiążącą.
Klejenie MO:
- Oddziaływania wiążące pomiędzy orbitami atomowymi są oddziaływaniami konstrukcyjnymi (w fazie).
- Wiążące się MO są mniej energochłonne niż orbity atomowe, które łączą się, aby je wytworzyć.
Antibonding MOs:
- Oddziaływania antywibondingowe pomiędzy orbitami atomowymi są oddziaływaniami destrukcyjnymi (poza fazami).
- Antybondingowe MOs mają wyższą energię niż orbity atomowe, które łączą się, aby je produkować.
Niewiążące się MO:
- Niewiążące się MO są wynikiem braku interakcji pomiędzy orbitami atomowymi z powodu braku kompatybilnych symetrii.
- Niewiążące się MO będą miały taką samą energię jak orbity atomowe jednego z atomów w cząsteczce.
HOMO i LUMO
Każda orbita molekularna ma swój własny poziom energii. Chemicy sortują MO według poziomów energii. Chemicy zakładają, że elektrony najpierw wypełnią MO o najniższym poziomie energetycznym. Na przykład, jeśli cząsteczka ma elektrony do wypełnienia 15 orbit, 15 MO z najniższym poziomem energii zostanie wypełnionych. 15 MO na liście będzie nazywane "najwyżej zajętą orbitą molekularną" (HOMO), a 16 MO na liście będzie "najniżej zajętą orbitą molekularną" (LUMO). Różnica w poziomie energii HOMO i LUMO nazywana jest luką pasmową. Luka pasmowa może czasami służyć jako miara pobudliwości cząsteczki: im mniejsza energia, tym łatwiej będzie ją pobudzić. Kiedy elektron jest wzbudzony, przeskakuje do niezamieszkałego MO. Na przykład, może to pomóc w zgadywaniu, czy coś da światło (luminescencja).

Pytania i odpowiedzi
P: Co to jest orbital molekularny?
A: Orbital molekularny (lub MO) to funkcja matematyczna, która opisuje falowe zachowanie elektronu w cząsteczce. Wyjaśnia ona, co dzieje się z elektronami, gdy atomy łączą się w cząsteczkę i może określić prawdopodobieństwo znalezienia elektronu w dowolnym określonym regionie.
P: Jak chemicy budują modele matematyczne orbitali molekularnych?
O: Chemicy zazwyczaj budują modele matematyczne orbitali molekularnych poprzez łączenie orbitali atomowych. Można również wykorzystać orbitale hybrydowe z każdego atomu cząsteczki lub inne orbitale molekularne z grup atomów. Komputery mogą pracować na tych funkcjach.
P: Co mechanika kwantowa ma wspólnego z badaniem cząsteczek?
O: Orbitale molekularne pozwalają chemikom stosować mechanikę kwantową do badania cząsteczek. Odpowiadają one na pytania, jak atomy w cząsteczkach trzymają się razem i dają wgląd we właściwości chemiczne i fizyczne.
P: Co to są diagramy orbitalne?
O: Diagramy orbitalne to wizualne przedstawienia, które wskazują, gdzie najprawdopodobniej znajdują się elektrony w atomie na podstawie jego różnych zaokrąglonych kształtów.
P: Jak działają orbitale hybrydowe?
O: Orbitale hybrydowe łączą różne typy orbitali atomowych w jeden nowy typ, który ma unikalne cechy w porównaniu do jego części składowych. Takie hybrydy są często wykorzystywane przy tworzeniu modeli matematycznych dla orbitali molekularnych.
P: Jak komputery mogą pomóc w badaniu MO?
O: Komputery mogą pomóc w badaniu MO, pracując nad ich funkcjami i zapewniając dokładniejsze przewidywania lub wyjaśnienia właściwości chemicznych i fizycznych w cząsteczkach.
Powiązane artykuły
Autor
AlegsaOnline.com Orbital molekularny — definicja, teoria i zastosowania w chemii kwantowej Leandro Alegsa
URL: https://pl.alegsaonline.com/art/65861
Źródła
- nobelprize.org : Nobelprize.org