Chemia obliczeniowa: teoria, metody i zastosowania
Przegląd chemii obliczeniowej: cele, metody (od mechaniki kwantowej po dynamikę molekularną), zastosowania w projektowaniu leków i materiałów, ograniczenia i narzędzia.
Chemia obliczeniowa to interdyscyplinarna gałąź nauki łącząca chemię i informatykę, która wykorzystuje modele matematyczne i obliczenia numeryczne do przewidywania właściwości układów chemicznych. W praktyce uzupełnia ona pracę eksperymentalną, pozwalając symulować struktury, energie i własności spektroskopowe zanim przeprowadzi się kosztowne badania laboratoryjne. Dla ogólnego omówienia dziedziny zobacz definicję i powiązania z informatyką.
Galeria obrazów
8 Obrazy


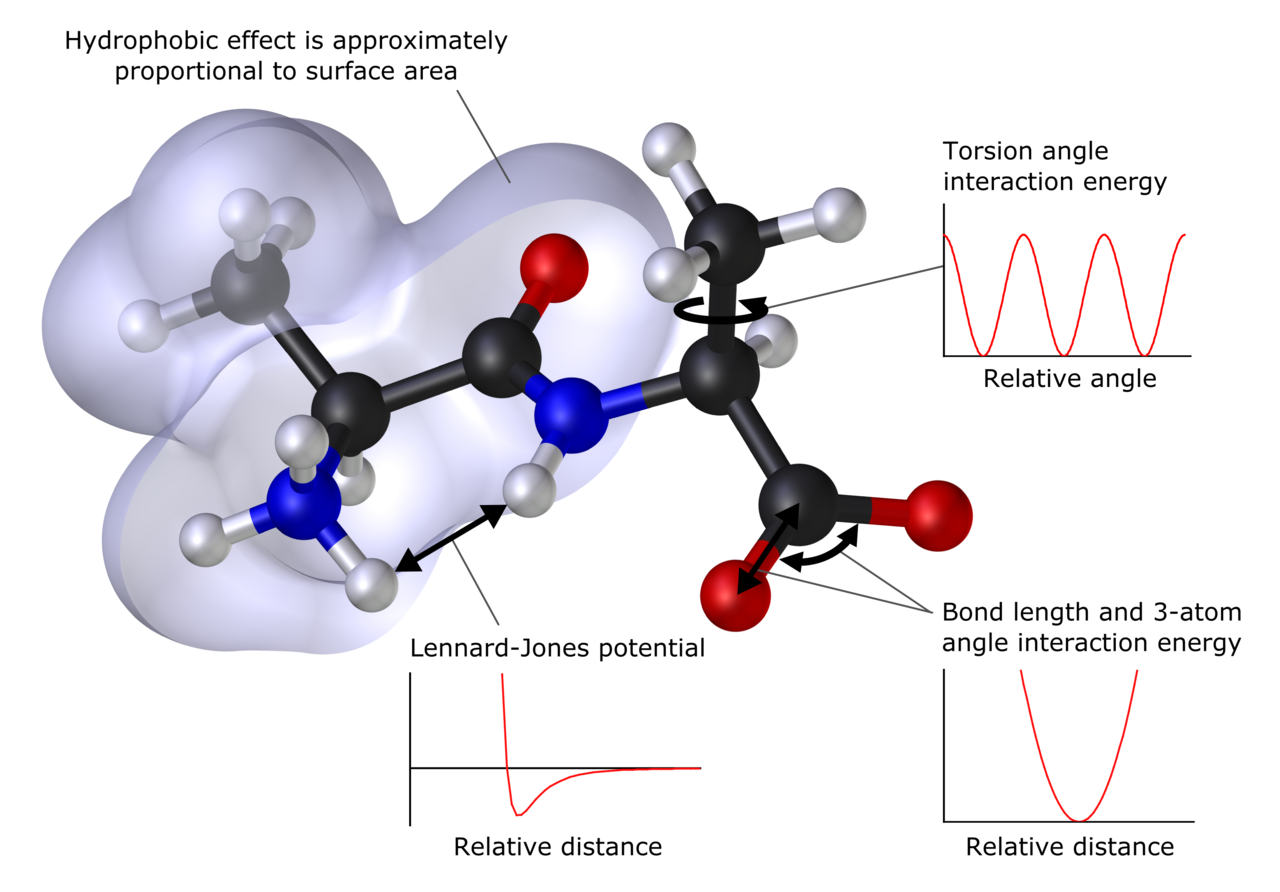


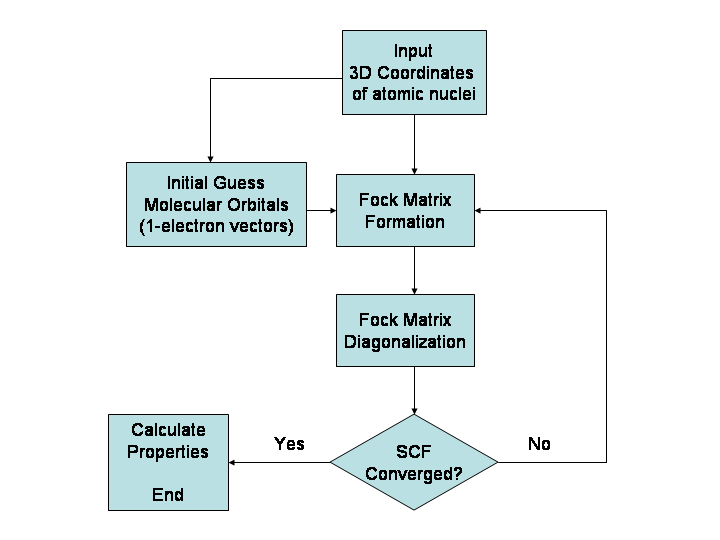
Co można przewidzieć i jakich wielkości dotyczy
Metody obliczeniowe potrafią oszacować rozmieszczenie atomów w cząsteczce, przewidzieć energie związku i interakcje międzycząsteczkowe, opisać rozkład ładunku i właściwości elektroniczne oraz wyliczyć momenty wielobiegunowe. Typowe cele obejmują strukturę pojedynczej cząsteczki, układy skondensowane takie jak ciała stałe, a także uzupełnianie wyników eksperymentów. Modelowanie może dotyczyć także przewidywania nowych zjawisk chemicznych przed ich obserwacją.
Główne klasy metod
- Metody pierwszych zasad (ab initio) i teoria funkcjonału gęstości — opis elektronów i energii energii, rozkładu elektronów.
- Dynamika molekularna — badanie zachowania układów w czasie przy użyciu sił między atomami; wyznaczanie częstości drgań i właściwości spektroskopowych.
- Metody przybliżone i półempiryczne — kompromis między dokładnością a kosztem obliczeniowym; stosowane dla większych systemów.
- Teorie reaktywności — szacowanie reaktywności, barier energetycznych i mechanizmów reakcji.
Historia i rozwój
Początki chemii obliczeniowej sięgają połowy XX wieku, kiedy rozwój komputerów umożliwił rozwiązywanie równań mechaniki kwantowej dla prostych molekuł. Z czasem algorytmy i moc obliczeniowa wzrosły, co zaowocowało rozszerzeniem zakresu problemów — od analizy drobnych cząsteczek po symulacje złożonych materiałów i układów biologicznych. Dalszy rozwój oprogramowania i sprzętu umożliwił stosowanie hybrydowych technik oraz integrację z bazami danych i narzędziami do projektowania.
Zastosowania praktyczne
Chemia obliczeniowa odgrywa ważną rolę w wielu dziedzinach: projektowaniu nowych leków, optymalizacji materiałów o pożądanych właściwościach, badaniu katalizy, a także w nanotechnologii. Dzięki symulacjom można przewidzieć pozycje atomów, dipole i wyższe momenty (dipole), wyznaczyć przekroje dla procesów zderzeniowych (zderzeń) oraz interpretować pomiary rezonansu czy spektroskopii.
Ograniczenia i praktyczne uwagi
Wydajność i dokładność metod zależy od wielkości układu i przyjętych przybliżeń. Bardzo dokładne obliczenia są zwykle wykonalne tylko dla małych molekuł; symulacje dużych systemów wymagają uproszczeń i więcej zasobów obliczeniowych, pamięci i przestrzeni dyskowej. Wybór odpowiedniej metody to kompromis między kosztami a potrzebną precyzją. Więcej informacji o narzędziach i praktykach znajduje się w materiałach wprowadzających o ciałach stałych oraz przeglądach technicznych zjawisk.
Gdzie szukać wiedzy i oprogramowania
- Podręczniki akademickie i kursy online — ogólne wprowadzenia i teoria.
- Pakiety obliczeniowe i biblioteki — dostępne narzędzia komercyjne i open source.
- Bazy danych struktur i wyników obliczeń — ułatwiają porównania i walidację wyników.
Dla osób zaczynających warto od kursów dotyczących podstaw mechaniki kwantowej i dynamiki molekularnej oraz od zasobów edukacyjnych i dokumentacji programów: przykładowe źródła i przewodniki znajdują się pod linkami do działów ogólnych dziedziny, informatyki i praktycznych poradników eksperymentów.
Uwaga: Chemia obliczeniowa to obszar dynamiczny — metody i narzędzia ciągle się rozwijają, a ich skuteczność zależy od konkretnego problemu badawczego.

Powiązane strony
- Bioinformatyka
- Mechanika statystyczna
Pytania i odpowiedzi
P: Co to jest chemia obliczeniowa?
O: Chemia obliczeniowa jest dziedziną chemii, która wykorzystuje informatykę do rozwiązywania problemów chemicznych. Za jej pomocą można obliczać struktury i właściwości cząsteczek i ciał stałych, przewidywać zjawiska chemiczne, które nie zostały jeszcze zaobserwowane, oraz projektować nowe leki i materiały.
P: Jakie rodzaje systemów bada chemia obliczeniowa?
O: Chemia obliczeniowa bada zarówno systemy statyczne, jak i dynamiczne. Układem może być pojedyncza cząsteczka, grupa cząsteczek lub ciało stałe.
P: Jakich rodzajów informacji może dostarczyć chemia obliczeniowa?
O: Chemia obliczeniowa może dostarczyć informacji takich jak struktura (pozycje atomów), energie bezwzględne i względne, rozkłady ładunków elektronicznych, dipole i wyższe momenty multipolowe, częstotliwości drgań, reaktywność lub inne wielkości spektroskopowe oraz przekroje poprzeczne na zderzenia z innymi cząsteczkami.
P: Jak dokładne są metody stosowane w chemii obliczeniowej?
O: Dokładność metod stosowanych w chemii obliczeniowej waha się od bardzo dokładnych do bardzo przybliżonych. Bardzo dokładne metody są zazwyczaj możliwe do zastosowania tylko w przypadku małych układów.
P: W jaki sposób chemia obliczeniowa uzupełnia dane eksperymentalne?
O: Chemia obliczeniowa zwykle uzupełnia informacje uzyskane w wyniku eksperymentów chemicznych. Można ją wykorzystać do przewidywania wyników, które nie zostały jeszcze zaobserwowane doświadczalnie.
P: Czy wielkość badanego systemu ma wpływ na to, jak dużo czasu trzeba poświęcić komputerowi?
O: Tak - wraz ze wzrostem wielkości badanego systemu rośnie ilość czasu komputerowego potrzebnego do analizy, a także zasoby takie jak pamięć i przestrzeń dyskowa potrzebne do przechowywania danych.
Powiązane artykuły
Autor
AlegsaOnline.com Chemia obliczeniowa: teoria, metody i zastosowania Leandro Alegsa
URL: https://pl.alegsaonline.com/art/22297