Reakcje cheletropowe: reakcje pericykliczne — definicja, mechanizm, przykłady
Reakcje cheletropowe — przystępna definicja, mechanizm i przykłady reakcji pericyklicznych z ilustracjami, mechanistycznym omówieniem i zastosowaniami w chemii.
Reakcje cheletropowe są szczególną odmianą reakcji pericyklicznej, w której z jednej strony tworzy się jednocześnie dwoje nowych wiązań chemicznych do tego samego atomu. Reakcja pericykliczna przebiega przez stan przejściowy o cyklicznym układzie atomów i odpowiadającym mu cyklicznym układzie oddziałujących orbitali — w jego trakcie następuje jednoczesna reorganizacja wiązań σ i π. W reakcjach cheletropowych oba nowe wiązania są utworzone z tego samego atomu jednego z reagentów, co odróżnia je od innych typów cykloaddycji.
Galeria obrazów
6 Obrazy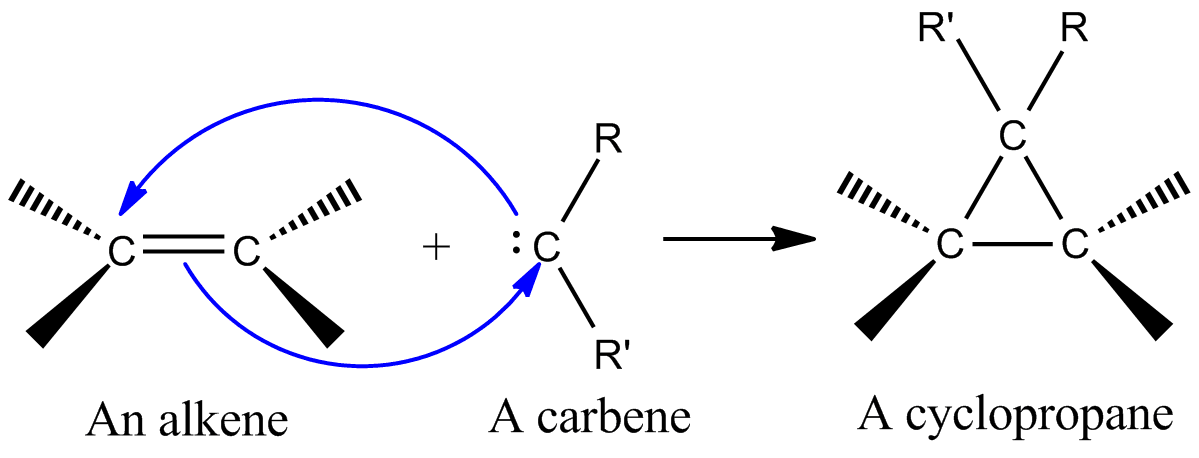




Charakterystyczne cechy
- Cheletropowe tworzenie/rozerwanie dwóch wiązań do jednego atomu reagentowego.
- Proces często przebiega w sposób skoordynowany (concerted), przez jednoczesne przesunięcie elektronów w zamkniętym cyklicznym stanie przejściowym.
- Wielu przykładom towarzyszy wydzielanie małej, stabilnej cząsteczki (np. CO, N2) — w takich przypadkach reakcję nazywa się czasem „wytrąceniem cheletropowym”. Siłą napędową jest często korzyść entropowa wynikająca z uwolnienia gazu.
- Stereochemia produktów jest zwykle ściśle kontrolowana przez mechanizm (stereospecyficzność), jeśli proces jest synchoronyczny i nie przebiega przez pośredniak rodnikowy.
Mechanizm orbitalny i reguły symetrii
Mechanizm cheletropowy mieści się w ramach teorii reakcji pericyklicznych. Kluczową rolę odgrywają tutaj wzajemne oddziaływania orbitali reagujących fragmentów (np. orbitali π układu wielokrotnego i orbitali karbenów, atomów z wolnymi parami itp.). Zastosowanie mają reguły symetrii Woodwarda–Hoffmanna: czy proces jest dozwolony termodynamicznie jako suprafacjalny czy wymaga przesunięć antarafacjalnych zależy od liczby elektronów biorących udział i układu orbitali. Dla prostych cheletropowych (np. dodawanie singletowych karbenów do alkenów) typowy jest skoncertowany, suprafacjalny przebieg, prowadzący do stereospecyficznych produktów.
Przykładowo, w przypadku addycji singletowego karbenu (:CHR) do alkenu, orbital p karbenu i jego wolna para współdziałają z układem π węgiel–węgiel, tworząc jednocześnie dwa wiązania C–C i powstaje cyklopropan. Jeśli karben jest w stanie tripletowym, reakcja często przebiega dwuetapowo przez pośredniak rodnikowy, co zmniejsza stereospecyficzność.
Przykłady reakcji cheletropowych
- Cyklopropanacja alkenów przez karbeny — klasyczny przykład: dichlorokarben (:CCl2) lub meteń (:CH2) dodaje się do wiązania C=C, tworząc cyklopropan; oba nowe wiązania tworzone są z tego samego atomu karbenu.
- Wytrącenia cheletropowe — reakcje, w których z układu odłączana jest mała cząsteczka, np. CO lub N2. W takich procesach zmiana entropii (uwolnienie gazu) jest często główną siłą napędową reakcji.
- Przykłady z tlenem i siarką — dodanie SO2 do dienów (prowadzące do związków typu sulfolenu) lub inne addycje, w których atom siarki/siarczanowy tworzy dwa nowe wiązania do jednego atomu reagentu, bywają klasyfikowane jako cheletropowe.
- Reakcje w chemii koordynacyjnej — tworzenie/rozerwanie wiązań między ligandami a metalami, gdzie jeden atom ligandu łączy się jednocześnie z dwoma centrami, może mieć charakter cheletropowy w sensie pericyklicznym.
Stereochemia i czynniki wpływające na przebieg
Główne czynniki decydujące o mechanizmie i stereochemii:
- Stan elektronowy reagentów (singlet vs triplet dla karbenów) — singlet sprzyja mechanizmowi skoordynowanemu, triplet — mechanizmowi dwuetapowemu przez rodniki.
- Symetria orbitali reagujących fragmentów — zgodność faz orbitali determinuje, czy przebieg jest dozwolony jako suprafacjalny lub wymagający antarafacjalności.
- Warunki termiczne — podwyższona temperatura sprzyja często procesom wytrącenia (eksergonicznym dzięki uwolnieniu gazu), natomiast niższe temperatury mogą faworyzować reakcje sterospecyficzne o niskiej energii aktywacji.
- Rozpuszczalnik i katalizatory — polarne rozpuszczalniki, katalizatory metaliczne lub fotochemia mogą zmieniać rodzaj karbenów/pośredników i tym samym mechanizm reakcji.
Znaczenie i zastosowania
Reakcje cheletropowe mają zastosowanie w syntezie organicznej (np. szybkie wprowadzanie trójczłonowych pierścieni przez cyklopropanację), w chemii materiałów oraz w procesach, gdzie kontrolowane wytrącenie małej cząsteczki jest wykorzystywane do napędu dalszych przemian. Rozumienie reguł orbitalnych pozwala chemikom projektować selektywne i stereospecyficzne warianty tych reakcji.
Podsumowanie
Reakcje cheletropowe to pericykliczne, często skoordynowane procesy, w których jeden atom jednego reagenta tworzy dwa wiązania z drugim reagentem. Mechanizm i stereochemia zależą od stanu elektronowego reagentów i symetrii orbitali; typowymi przykładami są cyklopropanacje karbenów i wytrącenia, w których uwalniany jest CO lub N2. Dzięki swojej specyfice i przewidywalnej stereochemii reakcje te są cennym narzędziem w syntezie organicznej.

Analiza teoretyczna
Ze względu na geometrię cząsteczek biorących udział w reakcjach cheletropowych, potwierdzają one szereg przewidywań poczynionych przez chemików teoretycznych. Reakcje cheletropowe potwierdzają zachowanie symetrii orbitali molekularnych.
W perycyklicznym stanie przejściowym, mała cząsteczka oddaje dwa elektrony do pierścienia. Dwie możliwe geometrie mogą wyjaśnić tę reakcję. Mała cząsteczka może zbliżać się w sposób liniowy lub nieliniowy. W podejściu liniowym, elektrony na orbitalu małej cząsteczki są skierowane bezpośrednio na układ π dużej cząsteczki. W przypadku podejścia nieliniowego, orbitale zbliżają się pod nieco innym kątem. Zdolność układu π do rotacji w miarę zbliżania się małej cząsteczki jest kluczowa dla tworzenia nowych wiązań. Kierunek rotacji będzie różny w zależności od tego, ile π-elektronów znajduje się w układzie. Poniżej przedstawiony jest schemat dwuelektronowego fragmentu zbliżającego się do czteroelektronowego π-systemu przy użyciu granicznych orbitali molekularnych. Rotacja będzie dezrotacyjna, jeżeli mała cząsteczka zbliża się liniowo i konrotacyjna, jeżeli cząsteczka zbliża się nieliniowo. Dysrotacyjne i konrotacyjne mówią, w jaki sposób rotują wiązania w układzie π. Dysrotacyjne oznaczają przeciwne kierunki, natomiast konrotacyjne - ten sam kierunek. Jest to również pokazane na poniższym schemacie.
Używając reguły Huckela, można stwierdzić, czy układ π jest aromatyczny czy antyaromatyczny. Jeśli jest aromatyczny, to w podejściu liniowym stosuje się ruch dyskretny, a w podejściu nieliniowym ruch konrotacyjny. Odwrotnie jest w przypadku układu antyaromatycznego. Podejścia liniowe będą miały ruch konrotacyjny, podczas gdy podejścia nieliniowe będą miały ruch dysrotacyjny.
Reakcje cheletropijne z udziałem SO2
Termodynamika
Kiedy dwutlenek siarki reaguje z butadienem i izoprenem, powstają dwa różne produkty. Mechanizm reakcji decyduje o tym, co powstanie. Możliwy jest zarówno produkt kinetyczny, jak i produkt termodynamiczny. Większa ilość produktu termodynamicznego powstaje niż kinetycznego. Produkt kinetyczny pochodzi z reakcji Dielsa-Aldera, podczas gdy reakcja cheletropowa tworzy bardziej stabilny termodynamicznie produkt. Szlak cheletropowy jest częściej wykorzystywany, ponieważ prowadzi do bardziej stabilnego adduktu z pierścieniem pięcioczłonowym. Poniższy schemat pokazuje różnicę pomiędzy tymi dwoma produktami. Ścieżka po lewej stronie pokazuje produkt termodynamiczny, podczas gdy ścieżka po prawej stronie pokazuje produkt kinetyczny. Suarez i Sordo wykazali to w 1995 roku. Pokazali to zarówno eksperymentalnie jak i za pomocą obliczeń gaussowskich.
Kinetyka
Jednym z przykładów jest reakcja cheletropowa 1,3-dienów z dwutlenkiem siarki. Chemicy dokładnie przyjrzeli się kinetyce tej reakcji. W 1976 r. Isaacs i Laila zmierzyli współczynniki kenetyczne dla addycji ditlenku siarki do pochodnych butadienu. Szybkość addycji była monitorowana w benzenie w temperaturze 30 °C z początkowym dwudziestokrotnym nadmiarem dieny. Do pomiaru zaniku SO2 użyto spektrofotometru do badania światła przy 320 nm. Reakcja wykazywała "pseudo kinetykę pierwszego rzędu". Chemicy stwierdzili, że grupy odbierające elektrony na dienie zmniejszają szybkość reakcji. Ponadto, na szybkość reakcji znaczący wpływ miały efekty steryczne 2-podstawników, przy czym bardziej nieporęczne grupy zwiększały szybkość reakcji. (Innymi słowy, im większa grupa atomów zwisających z drugiego atomu węgla, tym reakcja przebiegała szybciej). Autorzy przypisują to tendencji nieporęcznych grup do faworyzowania konformacji cisoidalnej dieny, która jest niezbędna w reakcji (patrz tabela poniżej). Dodatkowo, dla siedmiu dienów zmierzono szybkość reakcji w czterech temperaturach. Na podstawie tych pomiarów chemicy wykorzystali równanie Arrheniusa do obliczenia entalpii aktywacji (ΔH‡) i entropii aktywacji (ΔS‡) dla każdej reakcji. Była to jedna z pierwszych ważnych prób zbadania kenetyki reakcji cheletropowej.
| -Butadien | 104 k /min-1 (30 °C) (± 1-2%) absolutny | 104 k /min-1 (30 °C) (± 1-2%) względne | ΔH‡ /kcal mol-1 | ΔS‡ /cal mol-1 K-1 |
| 2-metylo | 1.83 | 1.00 | 14.9 | -15 |
| 2-etyl | 4.76 | 2.60 | 10.6 | -20 |
| 2-izopropyl | 13.0 | 7.38 | 12.5 | -17 |
| 2-tert-butyl | 38.2 | 20.8 | 10.0 | -19 |
| 2-neopentylu | 17.2 | 9.4 | 11.6 | -18 |
| 2-klor | 0.24 | 0.13 | N/A | N/A |
| 2-bromoetylu | 0.72 | 0.39 | N/A | N/A |
| 2-p-tolil | 24.7 | 13.5 | 10.4 | -19 |
| 2-fenyl | 17.3 | 9.45 | N/A | N/A |
| 2-(p-bromofenyl) | 9.07 | 4.96 | N/A | N/A |
| 2,3-dimetylo | 3.54 | 1.93 | 12.3 | -18 |
| cis-1-metylo | 0.18 | 0.10 | N/A | N/A |
| trans-1-metylo | 0.69 | 0.38 | N/A | N/A |
| 1,2-dimetylo-cykloheksan | 24.7 | 13.5 | 11.4 | -16 |
| 2-metylo-1,1,4,4-d4 | 1.96 | N/A | N/A | N/A |
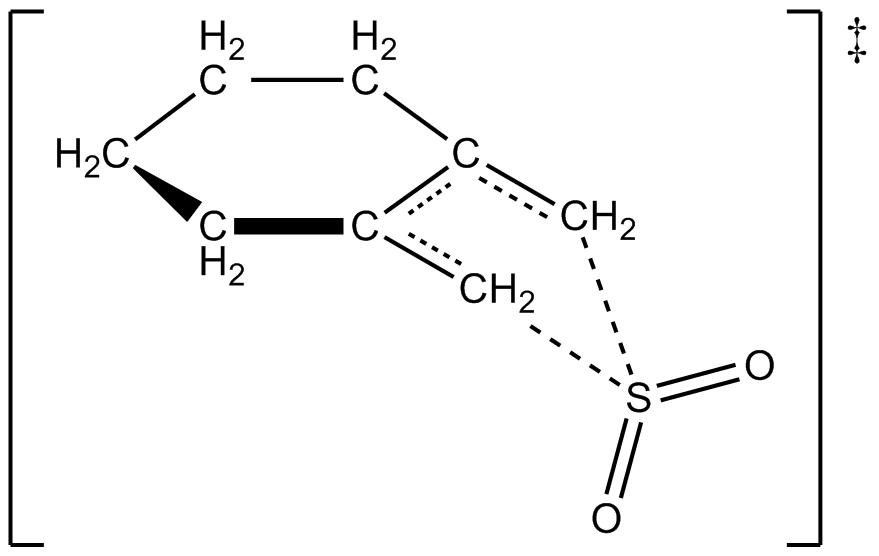
Monnat, Vogel i Sordo, w 2002 roku, zmierzyli kinetykę addycji dwutlenku siarki do 1,2-dimetyloidenocykloalkanów. Napisali oni, że reakcja 1,2-dimetyloidenocykloheksanu z dwutlenkiem siarki może dawać dwa różne produkty w zależności od warunków reakcji. Pod kontrolą kinetyczną (≤ -60 °C) reakcja tworzy odpowiednią sułtynę w reakcji hetero-Dielsa-Aldera, natomiast pod kontrolą termodynamiczną (≥ -40 °C) reakcja tworzy odpowiedni sulfolen w reakcji cheletropowej. Entalpia aktywacji dla reakcji hetero-Dielsa-Aldera jest o ok. 2 kcal/mol mniejsza niż dla odpowiadającej jej reakcji cheletropowej. Sulfolen jest o ok. 10 kcal/mol bardziej stabilny niż izometryczna sułtanka w roztworze CH2Cl2/SO2.
Autorom udało się doświadczalnie opracować prawo szybkości reakcji 1,2-dimetyloidenocykloheksanu z dwutlenkiem siarki w temperaturze 261,2 K, dając odpowiedni sulfolen. Reakcja była pierwszego rzędu w 1,2-dimetyloidenocykloheksanie, ale drugiego rzędu w ditlenku siarki (patrz poniżej). Potwierdziło to przewidywania chemików teoretycznych oparte na obliczeniach kwantowych wysokiego poziomu ab initio. Wykorzystując metody obliczeniowe, autorzy zaproponowali strukturę przejściową dla cheletropowej reakcji 1,2-dimetyloidenocykloheksanu z ditlenkiem siarki (patrz rysunek po prawej). Reakcja jest drugiego rzędu w dwutlenku siarki, ponieważ inna cząsteczka dwutlenku siarki prawdopodobnie wiąże się ze stanem przejściowym, aby pomóc w jego stabilizacji. Podobne wyniki uzyskano w badaniach przeprowadzonych w 1995 r. przez Suareza, Sordo i Sordo, którzy wykorzystali obliczenia ab initio do zbadania kinetycznej i termodynamicznej kontroli reakcji dwutlenku siarki z 1,3-dienami.
d [ 3 ] d t = k 2 [ 1 ] [ S O 2 ] 2 {{displaystyle {{frac {d[3]}{dt}}=k_{2}[1][SO_{2}]^{2}}} ![{\displaystyle {\frac {d[3]}{dt}}=k_{2}[1][SO_{2}]^{2}}](https://www.alegsaonline.com/image/a02f69dd0783356fbf24499f57d0df29cd264f16.svg)
Wpływ rozpuszczalnika
Zbadano kinetycznie wpływ rozpuszczalnika na przebieg cheletropowej reakcji 3,4-dimetylo-2,5-dihydrotiofen-1,1-ditlenku (rys. 1) w 14 rozpuszczalnikach. Stwierdzono, że stałe szybkości reakcji w przód i w tył, a także stałe równowagi są liniowo skorelowane ze skalą polarności rozpuszczalnika ET(30).
Reakcje prowadzono w temperaturze 120 °C i badano metodą spektroskopii 1H-NMR mieszaniny reakcyjnej. Stwierdzono, że szybkość reakcji w przód k1 maleje o współczynnik 4,5 przy przejściu z cykloheksanu do metanolu. Stwierdzono, że szybkość odwrotna k-1 wzrasta o współczynnik 53 przy przejściu z cykloheksanu do metanolu, a stała równowagi Keq maleje o współczynnik 140. Sugeruje się, że podczas procesu aktywacji zachodzi zmiana polarności, o czym świadczą zależności pomiędzy danymi równowagowymi i kinetycznymi. Autorzy twierdzą, że na przebieg reakcji wydaje się wpływać polarność rozpuszczalnika, a świadczyć o tym może zmiana momentów dipolowych przy przechodzeniu od reagenta do stanu przejściowego i produktu. Autorzy stwierdzają również, że na reakcję cheletropową nie wydaje się mieć wpływu ani kwasowość ani zasadowość rozpuszczalnika.
Wyniki tego badania skłaniają autorów do oczekiwania następujących zachowań:
1. Zmiana polarności rozpuszczalnika będzie miała mniejszy wpływ na szybkość niż równowaga.
2. Stałe szybkości będą charakteryzowały się przeciwnym wpływem na polaryzację: k1 będzie nieznacznie malało wraz ze wzrostem ET(30), a k-1 będzie rosło w tych samych warunkach.
3. Wpływ na k-1 będzie większy niż na k1.



Addycja karbenu do alkenów
Jedną z najważniejszych reakcji cheletropowych jest dodanie karbenu singletowego do alkenu w celu otrzymania cyklopropanu (patrz rysunek po lewej). Karben jest neutralną cząsteczką zawierającą węgiel dwuwartościowy z sześcioma elektronami w powłoce walencyjnej. Z tego powodu karbeny są wysoce reaktywnymi elektrofilami i powstają jako intermediaty reakcji. Karben singletowy zawiera pusty orbital p oraz orbital hybrydowy sp2, który posiada dwa elektrony. Karbeny singletowe przyłączają się stereospecyficznie do alkenów, a stereochemia alkenu jest zachowana w produkcie cyklopropanu. Mechanizm addycji karbenu do alkenu polega na zgodnej [2+1] cykloaddycji (patrz rysunek). Karbeny otrzymane z chloroformu lub bromoformu mogą być użyte do dodania CX2 do alkenu w celu otrzymania dihalocyklopropanu, podczas gdy odczynnik Simmonsa-Smitha dodaje CH2.
Oddziaływanie wypełnionego orbitalu karbenu z układem π alkenu tworzy układ czteroelektronowy i sprzyja podejściu nieliniowemu. Korzystne jest również mieszanie pustego orbitalu p karbenu z wypełnionym orbitalem π alkenu. Korzystne mieszanie zachodzi poprzez podejście nieliniowe (patrz rysunek 2 po prawej). Jednakże, podczas gdy teoria wyraźnie faworyzuje podejście nieliniowe, nie ma oczywistych implikacji doświadczalnych dla podejścia liniowego i nieliniowego.


Pytania i odpowiedzi
P: Co to jest reakcja cheletropowa?
O: Reakcja cheletropowa to rodzaj reakcji pericyklicznej, w której jeden atom na jednym z reagentów otrzymuje dwa nowe wiązania.
P: Co to jest reakcja pericykliczna?
O: Reakcja pericykliczna to taka, w której występuje stan przejściowy z cyklicznym układem atomów i związanym z nim cyklicznym układem oddziałujących orbitali, w którym następuje reorganizacja wiązań َ i ً.
P: Czym różnią się one od innych typów reakcji?
O: Reakcje cheletropowe są podklasą cykloaddycji i od innych typów reakcji odróżnia je to, że na jednym z reagentów oba nowe wiązania są tworzone do tego samego atomu.
P: Jakie są przykłady?
O: Przykładem mogą być "ekstrakcje cheletropowe", takie jak ta, w której pojedynczy atom w grupie karbonylowej kończy się w tlenku węgla.
P: Co napędza te reakcje?
O: Siłą napędową tych reakcji jest często korzyść entropowa z uwolnienia gazu (np. CO lub N2).
P: Czy rysunek 1 dotyczy reakcji cheletropowych? O: Tak, rysunek 1 przedstawia przykłady reakcji cheletropowych.
Powiązane artykuły
Autor
AlegsaOnline.com Reakcje cheletropowe: reakcje pericykliczne — definicja, mechanizm, przykłady Leandro Alegsa
URL: https://pl.alegsaonline.com/art/19133
Źródła
- doi.org : 10.1016/0040-4020(96)00279-7