Karbokation (kation węglowy) — definicja, budowa i reaktywność
Karbokation (kation węglowy) — definicja, budowa i reaktywność: wyjaśnienie hybrydyzacji sp2/sp3, stabilności i mechanizmów reakcji dla studentów i chemików.
Karbokation to jon z dodatnio naładowanym atomem węgla. Naładowany atom węgla w karbokationie posiada tzw. sekstet – to znaczy, że ma tylko sześć elektronów w zewnętrznej powłoce walencyjnej zamiast ośmiu, co stoi w sprzeczności z regułą oktetu. Z tego powodu karbokationy są zazwyczaj wysoko reaktywne: chętnie przyjmują elektrony lub reagują z nukleofilami, aby uzupełnić oktet i zneutralizować dodatni ładunek. Początkowa intuicja sugeruje hybrydyzację sp3 z pustym orbitalem sp3, jednak rzeczywista charakterystyka wielu karbokationów bliższa jest hybrydyzacji sp2 — obserwuje się geometrię trygonalną, a pusty orbital p leży prostopadle do płaszczyzny wiązań.
Galeria obrazów
2 Obrazy

Hybrydyzacja i geometria
W typowym, tzw. klasycznym karbokationie centralny atom węgla jest sp2-hybrydyzowany i ma płaską (trójkątną) geometrię. Trzy hybrydyzowane orbitale tworzą wiązania sigma z trzema sąsiednimi atomami, natomiast nieobsadzony orbital p pozostaje pusty i zawiera dodatni ładunek. To wyjaśnia, dlaczego karbokationy są silnymi elektrofilami — pusty orbital p łatwo przyjmuje parę elektronową od nukleofilów.
Stabilizacja karbokationów
- Stopień podstawienia: stabilność rośnie w kolejności: karbokationy trzeciorzędowe (tert.) > drugorzędowe > pierwotne > metylowy. Wynika to głównie z efektu hiperkonjugacji i indukcyjnego oddziaływania donorowego grup alkilowych.
- Hiperkonjugacja: przesunięcie gęstości elektronowej z sąsiednich wiązań C–H lub C–C do pustego orbitalu p stabilizuje jon. To ważny mechanizm, szczególnie dla karbokationów alkilowych.
- Rezonans (delokalizacja): karbokationy sprzężone z układami sprzężonymi (np. benzyliczne, allylowe) są znacząco stabilizowane przez rozdzielenie ładunku na kilka atomów węgla.
- Efekty indukcyjne i mezomerowe podstawnika: grupy donorowe (np. alkilowe) stabilizują, grupy przyciągające (np. halogeny) mogą destabilizować, chociaż halogeny dają też efekt rezonansowy w niektórych układach.
- Karbokationy nieklasyczne: istnieją struktury „mostkowe” (np. słynny 2-norbornylowy karbokation), w których ładunek jest częściowo oddzielony i stabilizowany przez nietypowe wiązania wielośrodkowe.
Klasyfikacja
- Karbokationy alifatyczne: metylowy, pierwotny, drugorzędowy, trzeciorzędowy.
- Karbokationy sprzężone: benzylowe, allylowe — stabilizowane rezonansowo.
- Karbokationy heterocykliczne i aromatyczne: występują specyficzne przypadki, np. kationy π w układach aromatycznych (jazda po regułach Hückla i aromatyczności).
Tworzenie karbokationów
- Heteroliza wiązań C–X: rozpad wiązania C–X (X = halogen, grupa odchodząca) prowadzi do powstania karbokationu i anionu — typowy krok w mechanizmach SN1 i E1.
- Protonowanie alkenów: przyłączenie H+ do wiązania wielokrotnego tworzy karbokation po stronie bardziej stabilnej (zasada Markownikowa).
- Oddziaływanie z elektrofikami i Lewisowymi kwasami: utlenianie, odłączenie ligandów silnie koordynowanych, czy kompleksowanie z kwasami Lewisa może generować karbokationy.
- Fotochemia i utlenianie jednorodzinne: w reakcjach fotolitycznych i w mechanizmach z rodnikami mogą powstawać pośrednio karbokationy.
Reaktywność i typowe reakcje
- Atak nukleofila: podstawowy los karbokationu — przyłączenie nukleofila do pustego orbitalu p prowadzi do powstania nowych wiązań i zobojętnienia ładunku.
- Eliminacje (E1): karbokationy mogą oddać proton z sąsiedniego węgla, tworząc alkene.
- Przestawienia (rearrangements): hydrydyczne lub alkilowe przesunięcia (np. przesunięcie 1,2-H lub 1,2-alkilu) często zachodzą, jeśli prowadzą do bardziej stabilnego karbokationu (np. z drugorzędowego do trzeciorzędowego).
- Reakcje kaskadowe: w syntezie organicznej wykorzystuje się zdolność karbokationów do inicjowania wielokrotnych przekształceń, np. cyklizacji elektrofilii.
Właściwości kinetyczne i termodynamiczne
Karbokationy bywają bardzo krótkotrwałe w warunkach normalnych, a tempo ich reakcji zależy zarówno od stabilności karbokationu, jak i stężenia oraz jakości nukleofila. W reakcjach SN1 szybkość zależy wyłącznie od szybkości tworzenia karbokationu (kroki rozdzielone) — im stabilniejszy jon, tym szybciej zachodzi etap wolnozależny.
Detekcja i badania
Karbokationy mogą być badane za pomocą spektroskopii NMR (np. charakterystyczne przesunięcia chemiczne), IR, UV–Vis oraz technik czasowo-rozdzielczych. W niektórych przypadkach stabilne karbokationy izolowano jako sole z mocnymi stabilizującymi anionami (np. barwne sole arenium), co pozwoliło na ich bezpośrednie badanie.
Znaczenie syntetyczne
Karbokationy odgrywają kluczową rolę w wielu reakcjach organicznych — od prostych zastąpień i eliminacji po złożone cyklizacje i syntezy naturalnych produktów. Kontrola powstawania karbokationów oraz ich migracji jest istotna przy planowaniu dróg syntezy i unikaniu niepożądanych przestawień.
Przykłady
- Kation tert-butylowy: bardzo stabilny ze względu na silną hiperkonjugację i efekt indukcyjny grup alkilowych.
- Kation benzylowy i allylowy: stabilizowane rezonansowo i często występujące jako pośredniki w reakcjach substitucji i addycji.
Podsumowując, karbokationy to elektrofilowe pośredniki z atomem węgla mającym niewypełniony oktet; mimo prostoty definicji ich właściwości i zachowanie zależą od złożonej kombinacji hybrydyzacji, stabilizacji przez podstawienie, rezonansu i innych efektów elektronowych, co czyni je centralnymi elementami chemii organicznej i syntezy.



Definicje
Karbokation był wcześniej często nazywany jonem węglowym, ale chemicy kwestionują jego dokładne znaczenie. W dzisiejszej chemii karbokationem jest każdy dodatnio naładowany atom węgla. Zasugerowano dwa specjalne typy: jony karbenowe są trójwartościowe, a jony węglowe są pięcio- lub sześciowartościowe. Podręczniki na poziomie uniwersyteckim omawiają karbokationy tylko tak, jakby były to jony karbenowe, lub omawiają karbokationy z przelotnym odniesieniem do starszego sformułowania jon karbenowy lub jony karbenowe i karbonowe. Jeden z podręczników do dnia dzisiejszego pozostaje przy starszej nazwie jonu węglowego dla jonu karbenowego i rezerwuje zwrot hiperwartościowy jon karbenowy dla CH5+.
Historia
W 1891 r. G. Merling poinformował, że dodał brom do tropylidenu (cykloheptatryny), a następnie podgrzał produkt, aby otrzymać krystaliczny, rozpuszczalny w wodzie materiał, C
7H
7Br. Nie zaproponował jego struktury, jednak Doering i Knox przekonująco wykazali, że jest to bromek tropylium (cykloheptatrienylium). Przewiduje się, że jon ten jest aromatyczny zgodnie z regułą Hückela.
W 1902 roku Norris i Kehrman niezależnie odkryli, że bezbarwny trifenylometanol daje głęboko żółte roztwory w stężonym kwasie siarkowym. Chlorek trifenylometylowy podobnie tworzył pomarańczowe kompleksy z chlorkami glinu i cyny. W 1902 r. Adolf von Baeyer rozpoznał solny charakter powstałych związków.
![]()
Zależność między kolorem a tworzeniem się soli nazwał halochromią, której doskonałym przykładem jest zieleń malachitowa.
Karbokationy są reaktywnymi intermediatami w wielu reakcjach organicznych. Idea ta, po raz pierwszy zaproponowana przez Juliusa Stieglitza w 1899 r., została rozwinięta przez Hansa Meerweina w jego badaniach z 1922 r. nad rearanżacją Wagnera-Meerweina. Stwierdzono również, że karbokationy biorą udział w reakcjiSN1, reakcji E1 oraz w reakcjach rearanżacji, takich jak przesunięcie Whitmore'a 1,2. Chemiczny establishment niechętnie akceptował pojęcie karbokacji i przez długi czas Journal of the American Chemical Society odrzucał artykuły, w których o nich wspominano.
Pierwsze widmo NMR stabilnej karbokationu w roztworze zostało opublikowane przez Doeringa i wsp. w 1958 roku. Był to jon heptametylobenzenoniowy, otrzymany w wyniku reakcji heksametylobenzenu z chlorkiem metylu i chlorkiem glinu. Stabilny kation 7-norbornadienylowy został otrzymany przez Story i wsp. w 1960 r. w reakcji chlorku norbornadienylowego z tetrafluoroboranem srebra w dwutlenku siarki w temperaturze -80 °C. Widmo NMR wykazało, że był to jon nieklasycznie zmostkowany (pierwszy zaobserwowany stabilny jon nieklasyczny).
W 1962 r. Olah bezpośrednio zaobserwował karbokation tert-butylowy za pomocą magnetycznego rezonansu jądrowego jako stabilny gatunek podczas rozpuszczania fluorku tert-butylu w kwasie magnezowym. NMR kationu norbornylu został po raz pierwszy opisany przez Schleyera et al. i wykazano, że ulega on protonowemu skramblingowi przez barierę przez Saundersa et al.
Właściwości
W chemii organicznej, karbokationy są często celem ataku nukleofilowego nukleofili, takich jak jony hydroksylowe (OH-) lub jony halogenowe.
Karbokationy są klasyfikowane jako pierwszorzędowe, drugorzędowe lub trzeciorzędowe w zależności od liczby atomów węgla związanych z węglem zjonizowanym. Karbokationy pierwszorzędowe mają jeden lub zero węgli przyłączonych do zjonizowanego węgla, karbokationy drugorzędowe mają dwa węgle przyłączone do zjonizowanego węgla, a karbokationy trzeciorzędowe mają trzy węgle przyłączone do zjonizowanego węgla.
Stabilność karbokationu wzrasta wraz z liczbą grup alkilowych przyłączonych do węgla przenoszącego ładunek. Karbokationy trzeciorzędowe są bardziej stabilne (i łatwiej się tworzą) niż karbokationy drugorzędowe; karbokationy pierwszorzędowe są bardzo niestabilne, ponieważ podczas gdy zjonizowane węgle wyższego rzędu są stabilizowane przez hiperkoniugację, niepodstawione (pierwszorzędowe) węgle nie są. Dlatego też reakcje takie jak reakcja SN1 i reakcja eliminacji E1 zwykle nie zachodzą, jeśli miałby powstać karbokation pierwszorzędowy. Wyjątkiem jest sytuacja, gdy obok zjonizowanego węgla znajduje się wiązanie podwójne węgiel-węgiel. Takie kationy jak kation allilowy CH2=CH-CH2+ i kation benzylowy C6H5-CH2+ są bardziej stabilne niż większość innych karbokationów. Cząsteczki, które mogą tworzyć karbokationy allilowe lub benzylowe są szczególnie reaktywne.
Karbokationy ulegają reakcjom rearanżacji z mniej stabilnych struktur do równie stabilnych lub bardziej stabilnych ze stałymi szybkości przekraczającymi 109/s. Fakt ten komplikuje drogę syntezy wielu związków. Na przykład, gdy 3-pentanol jest ogrzewany z wodnym HCl, początkowo utworzona 3-pentylowa karbokationacja rearanżuje się do statystycznej mieszaniny 3-pentylowej i 2-pentylowej. Kationy te reagują z jonem chlorkowym, tworząc około 1/3 3-chloropentanu i 2/3 2-chloropentanu.
Niektóre karbokationy, takie jak kation norbornylu, wykazują mniej lub bardziej symetryczne wiązanie trójcentrowe. Kationy tego rodzaju określa się mianem jonów nieklasycznych. Różnica energetyczna pomiędzy "klasycznymi" karbokationami a "nieklasycznymi" izomerami jest często bardzo mała, a energia aktywacji związana z przejściem pomiędzy strukturami "klasycznymi" i "nieklasycznymi" jest zazwyczaj niewielka, jeśli w ogóle występuje. Nieklasyczna" forma karbokationu 2-butylowego jest w istocie 2-butenem z protonem bezpośrednio powyżej centrum wiązania podwójnego węgiel-węgiel. "Nieklasyczne" karbokationy były kiedyś przedmiotem wielkich kontrowersji. Jednym z największych wkładów George'a Olah'a do chemii było rozwiązanie tej kontrowersji.

Karbokationy właściwe
Kationy cyklopropylokarbinylowe mogą być badane metodą NMR:
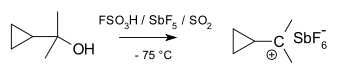
W widmie NMR pochodnej dimetylowej występują dwa nierównoważne sygnały dla dwóch grup metylowych wskazujące, że konformacja molekularna tego kationu nie jest prostopadła (jak w A), lecz dwupłaszczyznowa (jak w B) z pustym orbitalem p i układem pierścienia cyklopropylowego w tej samej płaszczyźnie:
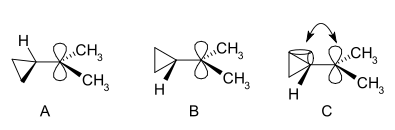
Z punktu widzenia teorii wiązań ugiętych, preferencja ta jest wyjaśniona przez założenie korzystnego nakładania się orbitali pomiędzy wypełnionymi wiązaniami ugiętymi cyklopropanu i pustym orbitalem p.
Pytania i odpowiedzi
P: Co to jest karbokracja?
A: Karbokracja to jon z dodatnio naładowanym atomem węgla.
P: Co to jest zewnętrzna powłoka walencyjna karbokationu?
A: Zewnętrzna powłoka karbokationu ma tylko sześć elektronów zamiast stabilnych ośmiu elektronów walencyjnych.
P: Dlaczego karbokationy są często reaktywne?
O: Karbokationy są często reaktywne, ponieważ starają się wypełnić oktet elektronów walencyjnych i odzyskać neutralny ładunek.
P: Jaka jest maksymalna stabilność atomów węgla?
O: Maksymalna stabilność atomów węgla jest osiągana, gdy mają one osiem elektronów walencyjnych.
P: Co to jest sekstet w chemii?
A: Sekstet to termin używany do opisania atomu węgla w karboksie, który ma tylko sześć elektronów w swojej zewnętrznej powłoce walencyjnej zamiast stabilnych ośmiu elektronów walencyjnych.
P: Jaka jest hybrydyzacja i geometria molekularna karbokationu?
O: Chociaż logika sugerowałaby, że karbokationy mają hybrydyzację sp3 z pustym orbitalem sp3 dającym ładunek dodatni, ich reaktywność bardziej przypomina hybrydyzację sp2 z trójkątną geometrią molekularną.
P: Co to jest reguła oktetu?
O: Reguła oktetu to zasada w chemii, która mówi, że atomy mają tendencję do tworzenia wiązań chemicznych z innymi atomami, które umożliwiają obu atomom posiadanie stabilnego zestawu ośmiu elektronów walencyjnych.
Tagi
Powiązane artykuły
Autor
AlegsaOnline.com Karbokation (kation węglowy) — definicja, budowa i reaktywność Leandro Alegsa
URL: https://pl.alegsaonline.com/art/16866
Źródła
- goldbook.iupac.org : carbonium ion
- iupac.org : PDF
- dx.doi.org : 10.1021/ja01641a027
- worldcat.org : 0096-4085
- dx.doi.org : 10.1016/0040-4020(58)88016-3
- dx.doi.org : 10.1021/ja01508a058
- dx.doi.org : 10.1021/ja01078a056
- dx.doi.org : 10.1021/ja01078a057
- nobelprize.org : George A. Olah - Nobel Lecture
- dx.doi.org : 10.1021/ja00713a080
- dx.doi.org : 10.1021/ja00950a026