Talasemia — co to jest? Przyczyny, objawy i leczenie
Talasemia — przyczyny, objawy i leczenie: zrozum przyczyny genetyczne, rozpoznaj symptomy, poznaj opcje terapeutyczne i profilaktykę nosicielstwa.
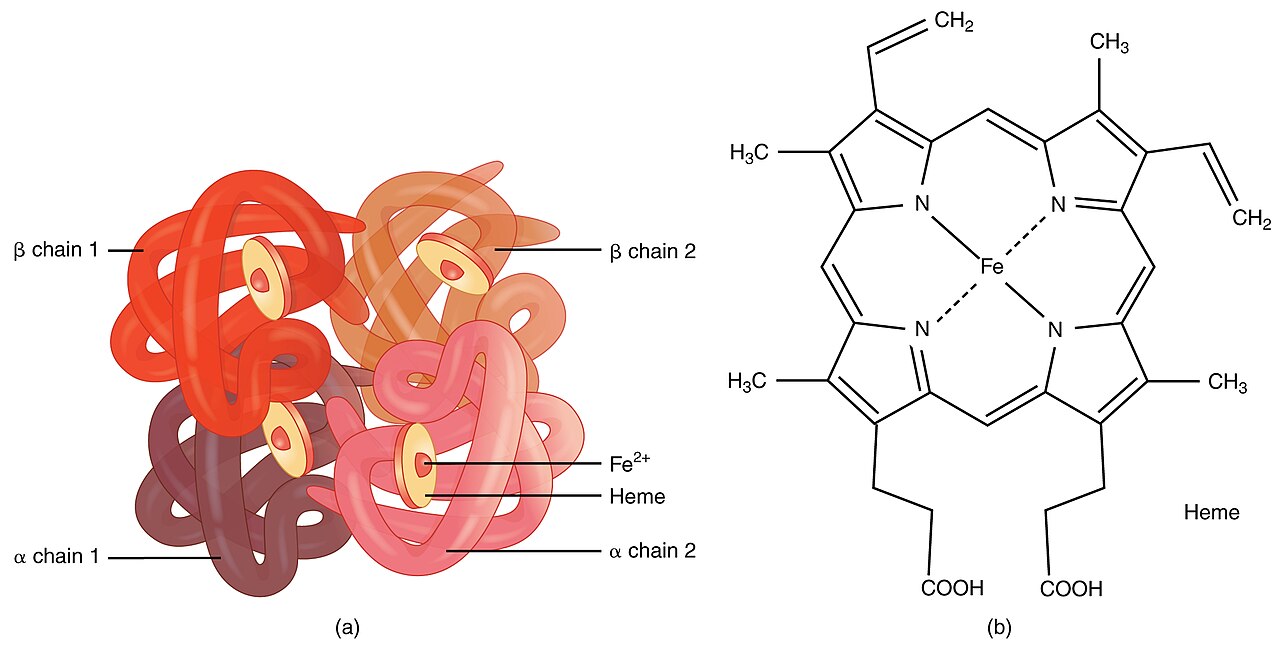
Talasemia (lub talasemia) jest genetycznym zaburzeniem krwi, które historycznie występowało najczęściej w populacjach z obszaru regionu śródziemnomorskiego, ale obecnie występuje na całym świecie w związku z migracjami. Choroba wynika z zaburzonej produkcji hemoglobiny — białka w czerwonych krwinkach odpowiedzialnego za transport tlenu.
Galeria obrazów
8 Obrazy





Przyczyny
Talasemia jest spowodowana przez zmutowane geny wpływające na sposób wytwarzania hemoglobiny. Osoby z talasemią wytwarzają mniej hemoglobiny i zwykle mniejszą liczbę krwinek czerwonych, co prowadzi do różnego stopnia anemii. Istnieją dwie główne grupy talasemii:
- Talasemia alfa — związana z mutacjami w genie(ach) kodujących łańcuchy alfa hemoglobiny.
- Talasemia beta — związana z mutacjami w genie kodującym łańcuch beta hemoglobiny.
W praktyce występuje wiele różnych wariantów mutacji, a każda z nich może dawać inny obraz kliniczny. Nosicielstwo (heterozygotyczność) jednego zmutowanego allelu często daje łagodniejszy obraz choroby i może zapewniać częściową ochronę przed malarią — zjawisko określane jako przewaga heterozygotyczna. To tłumaczy utrzymywanie się mutacji w niektórych populacjach na wysokim poziomie.
Dziedziczenie
Talasemia ma charakter dziedziczny. Jeśli oboje rodzice są nosicielami mutacji, istnieje ryzyko, że dziecko odziedziczy dwie kopie zmutowanego genu i będzie miało cięższą postać choroby. Stopień nasilenia zależy od tego, które i ile genów jest uszkodzonych.
Objawy
Obraz kliniczny talasemii zależy od typu i ciężkości mutacji. Najczęściej spotykane objawy to:
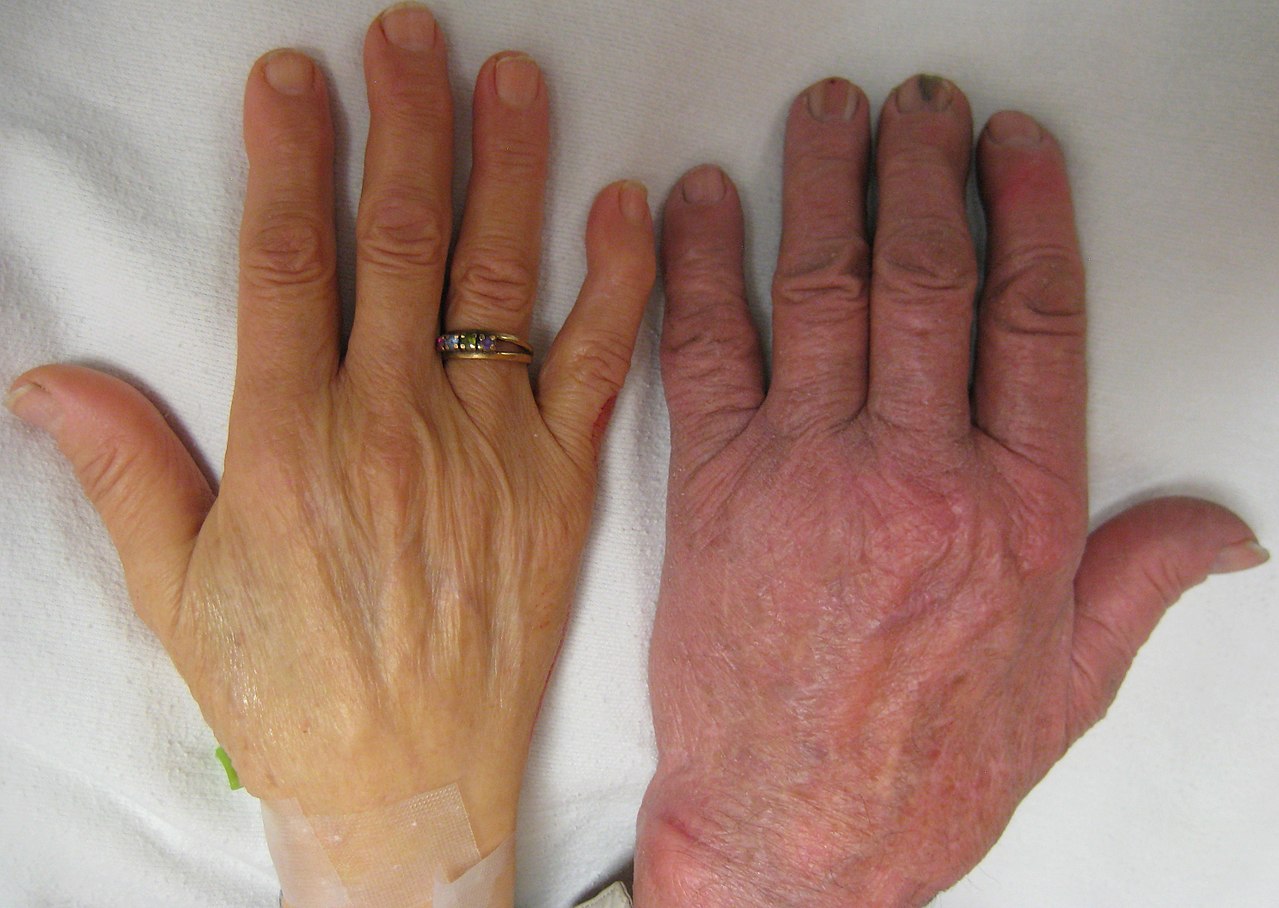
- zmęczenie, osłabienie, bladość skóry;
- krótkotrwałe duszności i szybkie męczenie się;
- opóźnienie wzrostu i rozwoju u dzieci;
- powiększenie śledziony (splenomegalia) i wątroby;
- deformacje kości twarzy i czaszki przy cięższych postaciach;
- żółtaczka i kamica żółciowa związana z nadmiernym rozpadem krwinek;
- objawy związane z przeciążeniem żelazem (zwłaszcza u osób leczonych regularnymi transfuzjami).
Nosiciele (osoby z tzw. cechą talasemii) często nie mają objawów lub mają jedynie łagodną anemię.
Rozpoznanie
Diagnostyka obejmuje badania krwi i badania molekularne:
- morfologia krwi (CBC) — wykrywa niedokrwistość i charakterystyczne zmiany w krwinkach;
- elektroforeza hemoglobiny — pozwala określić proporcje różnych typów hemoglobiny;
- badania genetyczne (analiza DNA) — identyfikacja konkretnej mutacji;
- dodatkowe badania biochemiczne (np. ferrytyna, transferyna) w celu oceny gospodarki żelazem.
Leczenie
Leczenie zależy od ciężkości choroby i obejmuje zarówno postępowanie doraźne, jak i długoterminowe:
- Transfuzje krwi — regularne podawanie koncentratów krwinek czerwonych u pacjentów z ciężką anemią; poprawiają jakość życia, ale prowadzą do akumulacji żelaza.
- Leczenie chelatujące żelazo — leki usuwające nadmiar żelaza z organizmu (np. deferoksamina, deferasiroks, deferipron) stosowane u osób po wielokrotnych transfuzjach lub z nadmiernym wchłanianiem żelaza.
- Usunięcie śledziony (splenektomia) — rozważane czasami u pacjentów z nadmiernym niszczeniem krwinek, lecz zwiększa ryzyko zakażeń.
- Przeszczep szpiku kostnego / przeszczep hematopoetyczny — istnieje możliwość leczenia pacjentów z talasemią za pomocą przeszczepów szpiku kostnego od zgodnych dawców. Metoda ta wymaga jednak kompatybilnego dawcy dopasowanego do HLA. Przeszczep może być potencjalnie leczniczy, ale wiąże się z ryzykiem powikłań i wymaga starannego doboru pacjentów.
- Terapie genowe — w ostatnich latach pojawiły się eksperymentalne i zatwierdzone formy terapii genowej/edytowania genów dla wybranych pacjentów z beta-talasemią; dostępność i wskazania zależą od kraju i ośrodka leczenia.
- Opieka wspomagająca — suplementy, rehabilitacja, monitorowanie powikłań sercowo-naczyniowych i metabolicznych oraz szczepienia (szczególnie po splenektomii).
Powikłania
Talasemia może prowadzić do poważnych powikłań, zwłaszcza przy cięższych postaciach lub przy niewłaściwym leczeniu:
- Zakażenia — zwłaszcza po splenektomii;
- przeciążenie żelazem — skutkujące uszkodzeniem serca, wątroby i gruczołów dokrewnych;
- deformacje kostne i zaburzenia wzrostu;
- choroby układu krążenia — niewydolność serca lub arytmie w wyniku gromadzenia żelaza w mięśniu sercowym.
Zapobieganie i doradztwo genetyczne
Dla par planujących ciążę ważne jest rozważenie badań przesiewowych i konsultacji genetycznej, szczególnie gdy w rodzinie występowała talasemia lub pochodzenie geograficzne wiąże się z wyższym ryzykiem. Dostępne są badania prenatane (np. analiza DNA z płynu owodniowego lub biopsji kosmówki) umożliwiające ustalenie, czy płód jest dotknięty chorobą.
Rokowanie i jakość życia
Rokowanie zależy od typu talasemii i dostępu do leczenia. Osoby z łagodnymi formami często prowadzą normalne życie. Pacjenci z ciężkimi postaciami, którzy otrzymują regularne transfuzje i skuteczne leczenie chelatujące, mogą mieć znacznie lepsze rokowanie i dłuższe życie niż w przeszłości. Przeszczepienie komórek macierzystych może być szansą na wyleczenie u wybranych pacjentów.
Gdzie szukać pomocy
- hematolog — podstawowy specjalista prowadzący leczenie talasemii;
- ośrodki referencyjne zajmujące się chorobami hemoglobinopatii;
- poradnictwo genetyczne przed planowaną ciążą;
- stowarzyszenia pacjentów i grupy wsparcia — pomoc w praktycznych i psychospołecznych aspektach życia z chorobą.
Jeśli podejrzewasz u siebie lub u bliskich talasemię (np. na podstawie przewlekłej anemii lub wyników badań), zgłoś się do lekarza POZ lub hematologa. Wczesna diagnostyka i właściwe leczenie znacząco poprawiają jakość życia i zmniejszają ryzyko powikłań.
Pytania i odpowiedzi
P: Co to jest talasemia?
A: Talasemia to genetyczne zaburzenie krwi, które powstało w rejonie Morza Śródziemnego. Choroba ta jest spowodowana osłabieniem i zniszczeniem czerwonych krwinek z powodu zmutowanych genów, które wpływają na sposób wytwarzania hemoglobiny przez organizm.
P: Jakie są niektóre powikłania związane z talasemią?
O: Powikłania związane z talasemią mogą obejmować zapalenie płuc, przeciążenie żelazem, deformacje kości i choroby układu krążenia.
P: W jaki sposób talasemia chroni przed malarią?
O: Nosiciele talasemii mają selektywną przewagę w przeżyciu dla nosicieli (tzw. przewagę heterozygotyczną), która pomaga utrzymać mutację w populacjach znacznie przewyższających wskaźnik mutacji. Dzięki temu chronią się przed malarią, która jest lub była powszechna w regionach, w których ta cecha występuje.
P: Czy istnieją różne wersje talasemii?
O: Tak, istnieje wiele różnych wersji talasemii, a każda z nich jest spowodowana mutacją w innym miejscu genomu. Przypomina to inną chorobę genetyczną dotyczącą hemoglobiny, chorobę sierpowatą.
P: Czy możliwe jest wyleczenie pacjentów z talasemią?
O: Tak, możliwe jest wyleczenie pacjentów z talasemią poprzez przeszczep szpiku kostnego od zgodnych dawców, którzy mają zgodnego w układzie HLA.
Powiązane artykuły
Autor
AlegsaOnline.com Talasemia — co to jest? Przyczyny, objawy i leczenie Leandro Alegsa
URL: https://pl.alegsaonline.com/art/97356
Źródła
- accessmedicine.com : accessmedicine.com/content.aspx?aID=6123722
- mayoclinic.com : mayoclinic.com/health/thalassemia/DS00905/DSECTION=complications
- bloodjournal.hematologylibrary.org : HLA-matched sibling bone marrow transplantation for β-thalassemia major