Zespół Patau (trisomia 13) — przyczyny, objawy i częstość występowania
Zespół Patau (trisomia 13) — przyczyny, objawy i częstość występowania. Dowiedz się o diagnostyce, ryzyku, przebiegu i statystykach tej rzadkiej choroby genetycznej.
Zespół Patau, znany również jako trisomia 13 (czasami nazywana trisomią D), to wada genetyczna wynikająca z obecności dodatkowej kopii chromosomu 13. Jest to zwykle efekt błędu podczas mejozy, prowadzącego do tzw. pełnej trisomii 13, choć możliwe są też postacie mozaikowe oraz przypadki związane z translokacją Robertsonowską. W praktyce oznacza to, że komórki dziecka mają trzy kopie chromosomu 13 zamiast dwóch, co zaburza prawidłowy rozwój. chromosomami
Galeria obrazów
4 Obrazy


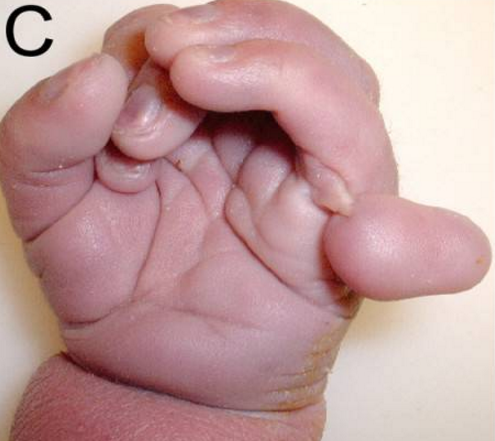
Przyczyny i rodzaje
- Pełna trisomia 13 – najczęstsza forma, zwykle powstaje na skutek nondysjunkcji chromosomów podczas gametogenezy (mejozy) i jest zwykle zmianą de novo.
- Mozai cyzm – tylko część komórek ma dodatkowy chromosom 13; przebieg może być łagodniejszy i bardzo zmienny.
- Translokacja Robertsonowska – dodatkowy materiał chromosomowy 13 może być przeniesiony na inny chromosom; wtedy rodzic może być nosicielem translokacji i istnieje zwiększone ryzyko nawrotu w kolejnych ciążach.
- Ryzyko wystąpienia zaburzenia wzrasta wraz z wiekiem matki; w badaniach średni wiek matek dzieci z trisomią 13 wynosił około 31 lat.
Objawy i cechy kliniczne
Spektrum objawów jest szerokie i obejmuje liczne wrodzone wady rozwojowe. Do najczęstszych należą:
- ciężkie zaburzenia rozwoju mózgu, w tym holoprosencefalia (nieprawidłowy podział przodomózgowia),
- małogłowie lub mikrocefalia,
- zaburzenia twarzoczaszki: małe oczy (microftalmia), wadliwe powieki, rozszczep wargi i podniebienia, nisko osadzone uszy, deformacje nosa,
- polidaktylia (dodatkowe palce),
- ciężkie wady serca (np. ubytki przegrody),
- wady nerek, problemy z układem oddechowym i pokarmowym, napięcie mięśniowe i ciężkie ograniczenia rozwojowe.
Diagnostyka
- W przesiewie prenatalnym pomocne są badania ultrasonograficzne (możliwe wykrycie wad anatomicznych, małej głowy, holoprosencefalii, wad serca, polidaktylii) oraz badania przesiewowe krwi i testy nieswoiste (np. ocena przezierności karkowej).
- Wysoką czułość ma nieinwazyjne badanie prenatalne z analizą wolnego DNA płodowego (NIPT/cfDNA), które sugeruje ryzyko trisomii.
- Rozstrzygające są badania diagnostyczne: kariotyp z pobranych komórek z biopsji kosmówki (CVS) lub z płynu owodniowego (amniopunkcja) oraz badania cytogenetyczne lub molekularne potwierdzające dodatkową kopię chromosomu 13.
- Po urodzeniu rozpoznanie potwierdza się badaniem kariotypu dziecka.
Leczenie i opieka
Nie ma leczenia przyczynowego usuwającego dodatkowy chromosom. Opieka jest objawowa i wielospecjalistyczna:
- leczenie chirurgiczne wad anatomicznych (np. operacje wad serca, korekcje rozszczepu), jeśli stan dziecka i decyzje rodzinne na to pozwalają,
- leczenie objawowe i wspomagające (terapia oddechowa, żywieniowa, fizjoterapia),
- opieka paliatywna i wsparcie rodzin, planowanie opieki w porozumieniu z rodzicami i zespołem medycznym.
Rokowanie
Rokowanie jest zwykle poważne. Większość noworodków z zespołem Patau ma ciężkie wady, co prowadzi do wysokiej śmiertelności we wczesnym okresie życia. Wielu niemowląt umiera w pierwszym miesiącu życia; odsetek przeżywalności do 1. roku jest niski, chociaż niewielka liczba dzieci przeżywa dłużej (przebieg zależy od ciężkości wad i postaci choroby, np. mozaikowej).
Częstość występowania
Jest to najrzadsza z trzech powszechnych trisomii obok zespołu Downa i zespołu Edwardsa. Szacunki częstości różnią się w zależności od populacji i metodologii badań; w literaturze podawane są wartości rzędu kilku do kilkunastu przypadków na 100 000 żywych urodzeń – w przybliżeniu mówi się o występowaniu około 1 na 10 000–25 000 żywych urodzeń, a w jednym ze źródeł spotyka się informację, że zespół Patau występuje u około jednego na 25 000 żywych urodzeń.
Ryzyko nawrotu i porady genetyczne
- W przypadkach pełnej trisomii powstawanie zmiany jest zwykle losowe, a ryzyko nawrotu w kolejnych ciążach jest niskie.
- Jeżeli u dziecka stwierdzono translokację Robertsonowską, konieczne jest badanie kariotypu rodziców — nosicielstwo translokacji u jednego z rodziców zwiększa ryzyko nawrotu w kolejnych ciążach.
- Zaleca się konsultację z genetykiem oraz badania kariotypu rodziców przed planowaniem kolejnej ciąży, aby oszacować indywidualne ryzyko i omówić dostępne opcje diagnostyczne.
Wsparcie dla rodzin
Diagnoza zespołu Patau jest trudnym doświadczeniem emocjonalnym i medycznym. Ważne jest zapewnienie rodzinie rzetelnej informacji, dostępu do poradnictwa genetycznego, opieki psychologicznej oraz wsparcia społecznego. Decyzje dotyczące leczenia i opieki powinny być podejmowane indywidualnie, z uwzględnieniem stanu dziecka oraz wartości i życzeń rodziny.
Autor
AlegsaOnline.com Zespół Patau (trisomia 13) — przyczyny, objawy i częstość występowania Leandro Alegsa
URL: https://pl.alegsaonline.com/art/74989
Źródła
- wrongdiagnosis.com : "Prevalence and Incidence of Patau syndrome"
- miscarriage.about.com : miscarriage.about.com