Trisomia 18 (zespół Edwardsa) — definicja, objawy, przyczyny i rokowania
Trisomia 18 (zespół Edwardsa): definicja, objawy, przyczyny i rokowania — diagnostyka, przebieg, szanse przeżycia i wsparcie dla rodzin. Dowiedz się więcej.
Trisomia 18, znana również jako zespół Edwardsa, jest trisomią. Jest to zaburzenie genetyczne. Ludzie z trisomią 18 mają trzy kopie chromosomu 18 zamiast dwóch — dodatkowy materiał chromosomowy zaburza rozwój płodowy i prowadzi do wielu wad wrodzonych. Nazwa zespołu pochodzi od nazwiska Johna H. Edwardsa, który po raz pierwszy opisał go w 1960 roku. Jest to druga najczęstsza trisomia autosomalna, po zespole Downa. Zespół Edwardsa często prowadzi do obumarcia płodu lub bardzo wczesnego zgonu po urodzeniu.
Galeria obrazów
3 Obrazy

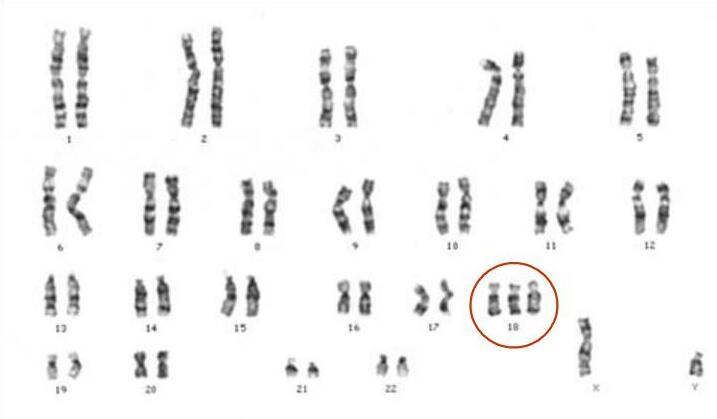
Częstość występowania i czynniki ryzyka
Szacuje się, że problem ten dotyczy około 1 na 2 500–3 000 żywych urodzeń (różne źródła podają nieco inne wartości). Częstość występowania wzrasta wraz z wiekiem matki — ryzyko nondysjunkcji podczas podziału komórek rozrodczych jest wyższe u starszych matek. Większość przypadków wynika z przypadkowego błędu w mejozie (niemutującego przekazania dodatkowego chromosomu), ale rzadziej trisomia może być efektem translokacji chromosomowej lub mozaicyzmu.
Przyczyny
- Nondysjunkcja w czasie mejozy: najczęstsza przyczyna — komórka jajowa lub plemnik wnosi dodatkowy chromosom 18.
- Mozaicyzm: część komórek organizmu ma prawidłowy zestaw chromosomów, a część ma trisomię — objawy bywają łagodniejsze (mozaikowy zespół Edwardsa).
- Translokacje: rzadziej dodatkowy materiał chromosomowy może być wynikiem translokacji równowagi u jednego z rodziców; wtedy ryzyko nawrotu w kolejnych ciążach jest wyższe.
Typowe cechy i objawy
U dzieci z trisomią 18 występuje zestaw charakterystycznych cech i wad anatomicznych oraz zaburzeń rozwojowych. Najczęściej obserwowane:
- niska masa urodzeniowa i trudności z przyrostem masy ciała,
- mała głowa (microcephalia), mała żuchwa (micrognathia), nisko osadzone uszy,
- ręce ze zgiętymi i nakładającymi się palcami (często palec wskazujący nakłada się na środkowy),
- stopy „rocker‑bottom” (zaokrąglone podeszwy stóp),
- wady serca — najczęściej ubytki przegrody (VSD), przetrwały przewód tętniczy (PDA) i inne nieprawidłowości,
- wady nerek i układu moczowego,
- zaburzenia przewodu pokarmowego, w tym atrezje lub przepukliny,
- ciężkie opóźnienie rozwoju psychoruchowego i znacząca niepełnosprawność intelektualna,
- częste problemy z oddychaniem, trudności z karmieniem i infekcje.
Diagnostyka
Rozpoznanie może być podejrzewane już w badaniach prenatalnych i potwierdzane po urodzeniu.
- Badania przesiewowe w ciąży: badanie ultrasonograficzne (nieprawidłowości anatomiczne, mała masa płodu, zaburzenia budowy serca), pomiar przezierności karkowej, oraz nowoczesne testy typu cfDNA (NIPT) wykrywające nieprawidłowości chromosomowe z krwi matki — te testy są przesiewowe, nie diagnostyczne.
- Diagnostyka inwazyjna: pobranie kosmówki (CVS) lub amniopunkcja pozwala na badanie kariotypu płodu i daje potwierdzenie rozpoznania.
- Po urodzeniu: rozpoznanie opiera się na obrazie klinicznym i potwierdzeniu genetycznym (kariotyp z krwi). W przypadku podejrzenia mozaicyzmu warto zbadać materiał z różnych tkanek.
Leczenie i opieka
Nie ma leczenia przyczynowego usuwającego dodatkowy chromosom. Opieka jest głównie wspomagająca i objawowa oraz dostosowana do indywidualnych potrzeb dziecka:
- leczenie wad serca — w niektórych przypadkach możliwe są interwencje chirurgiczne lub zabiegi kardiologiczne,
- wsparcie żywieniowe (np. sondy do karmienia), terapia logopedyczna i rehabilitacja,
- leczenie wad układu moczowego i innych wad wrodzonych w miarę możliwości medycznych,
- leczenie infekcji i problemów oddechowych,
- opieka paliatywna i wsparcie dla rodziny — ważne decyzje dotyczące zakresu leczenia podejmowane są indywidualnie, z uwzględnieniem prognozy i życzeń rodziców,
- konsultacje wielospecjalistyczne (genetyk, kardiolog dziecięcy, neonatolog, nefrolog, neurolog, rehabilitant).
Rokowania
Zespół Edwardsa wiąże się z bardzo niskim wskaźnikiem przeżywalności. Około 95% zliczanych płodów z tym zespołem obumiera przed urodzeniem lub w tej samej ciąży; spośród dzieci urodzonych żywo znaczna część umiera w ciągu pierwszych miesięcy życia. W literaturze medycznej podawane są następujące orientacyjne dane:
- około połowa dzieci urodzonych z tym schorzeniem przeżywa pierwszy miesiąc życia,
- tylko 5–10% przeżywa pierwszy rok,
- mniejszy odsetek (ok. 1%) może dożyć wieku 10 lat — najczęściej dotyczy to pacjentów z mniej wyrażonym mozaicyzmem lub łagodniejszym przebiegiem choroby.
Rokowanie zależy od ciężkości wad współistniejących (zwłaszcza serca i układu oddechowego) oraz od tego, czy występuje mozaicyzm.
Zapobieganie i doradztwo genetyczne
Nie ma sposobu, by zapobiec nondysjunkcji chromosomowej w danej ciąży. Dla par, w których stwierdzono trisomię 18 u dziecka, rekomendowane jest:
- skonsultowanie się z genetykiem klinicznym w celu omówienia przyczyny i ryzyka nawrotu,
- badanie kariotypu rodziców — w przypadku wykrycia zrównoważonej translokacji u jednego z rodziców ryzyko nawrotu jest istotnie wyższe i wymaga szczegółowego omówienia opcji reprodukcyjnych,
- rozważenie badań przesiewowych i diagnostycznych w kolejnych ciążach (NIPT, USG, CVS/amniopunkcja),
- wsparcie psychologiczne i informacje o dostępnych formach opieki i decyzjach medycznych.
Uwagi praktyczne dla rodziców
Diagnoza trisomii 18 jest trudnym doświadczeniem. Rodziny potrzebują informacji, empatycznego wsparcia oraz indywidualnego planu opieki dla dziecka, obejmującego dostęp do specjalistów, opiekę paliatywną i pomoc psychospołeczną. Decyzje dotyczące leczenia inwazyjnego należy podejmować po omówieniu możliwych korzyści i ryzyk oraz z uwzględnieniem wartości i preferencji rodziny.
Pytania i odpowiedzi
P: Co to jest trisomia 18?
A: Trisomia 18, znana również jako zespół Edwardsa, to choroba genetyczna, w której osoby dotknięte chorobą mają trzy kopie chromosomu 18 zamiast normalnych dwóch.
P: Kim jest John H. Edwards?
O: John H. Edwards jest osobą, od której pochodzi nazwa Trisomia 18. Po raz pierwszy opisał on ten zespół w 1960 roku.
P: Jak często występuje Trisomia 18?
A: Trisomia 18 występuje u około jednego na 3 000 żywych urodzeń.
P: Jakie są częste komplikacje zdrowotne związane z Trisomią 18?
O: U osób z trisomią 18 często występują nieprawidłowości w budowie serca, wady rozwojowe nerek i inne zaburzenia organów wewnętrznych.
P: Jaki jest wskaźnik przeżywalności dzieci z trisomią 18?
O: Około 95% dzieci z trisomią 18 umiera przed urodzeniem. Około połowa dzieci urodzonych z tą wadą osiągnie wiek dwóch miesięcy, a tylko 5-10% przeżyje rok. Mediana długości życia wynosi od pięciu do piętnastu dni.
P: Jaki odsetek dzieci urodzonych z trisomią 18 dożywa 10 lat?
O: Jeden procent dzieci urodzonych z trisomią 18 dożywa dziesiątego roku życia, zazwyczaj w przypadku mniej ciężkiego mozaikowego zespołu Edwardsa.
P: Czy częstość występowania trisomii 18 zwiększa się wraz z wiekiem matki?
O: Tak, częstość występowania trisomii 18 wzrasta wraz z wiekiem matki.
Powiązane artykuły
Autor
AlegsaOnline.com Trisomia 18 (zespół Edwardsa) — definicja, objawy, przyczyny i rokowania Leandro Alegsa
URL: https://pl.alegsaonline.com/art/30307
Źródła
- whonamedit.com : "Edwards syndrome (John Hilton Edwards)"
- nlm.nih.gov : nlm.nih.gov/MEDLINEPLUS/ency/article/001661.htm
- books.google.com : Fetal Medicine: Basic Science and Clinical Practice
- findarticles.com : findarticles.com
- emedicine.com : "Introduction to Trisomy 18"